

Phosphorylation barcodes direct biased chemokine signaling at CXCR3
- PMID: 37030291
- PMCID: PMC10147449
- DOI: 10.1016/j.chembiol.2023.03.006
Phosphorylation barcodes direct biased chemokine signaling at CXCR3
Abstract
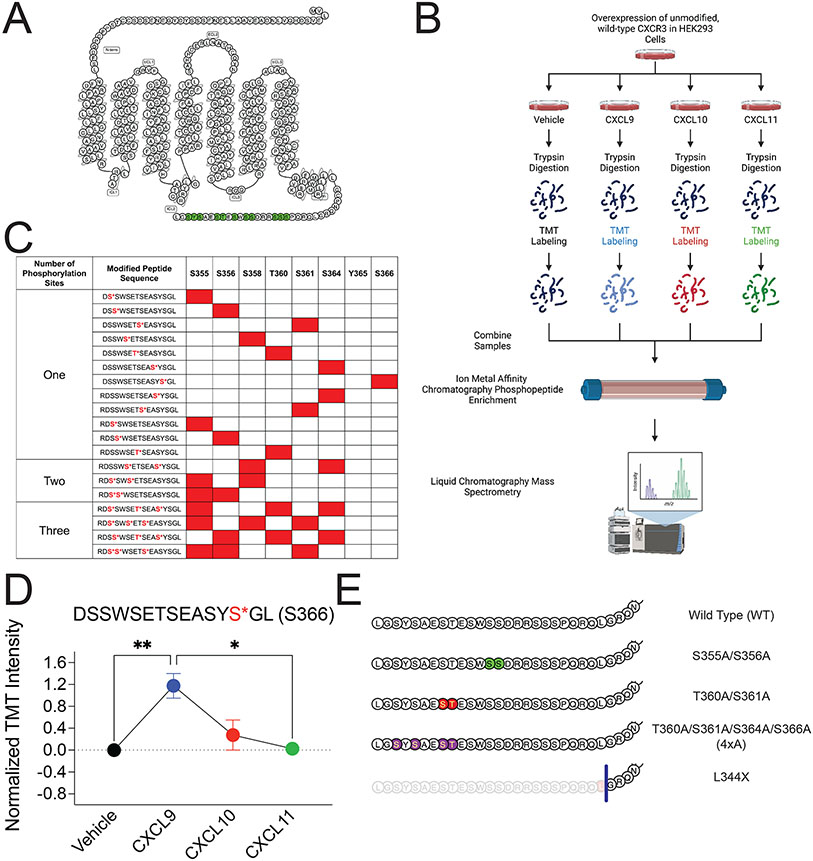
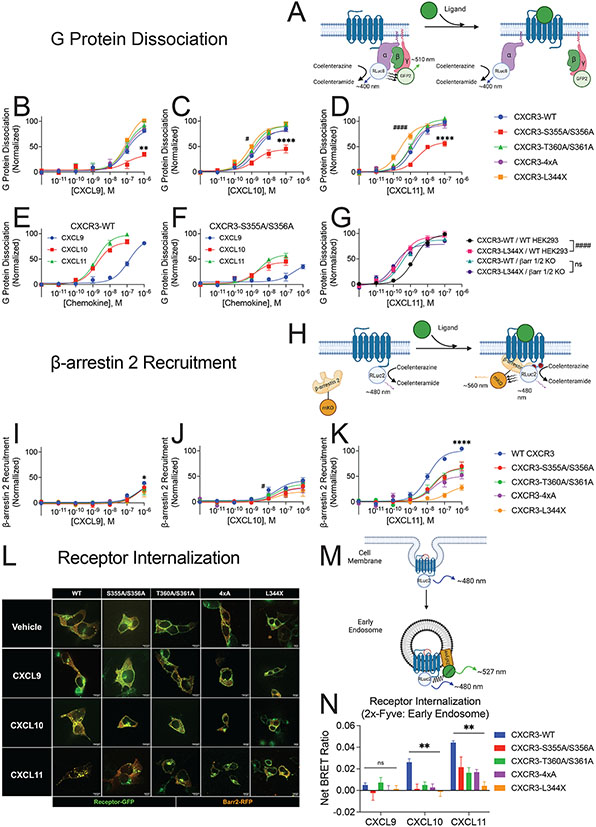
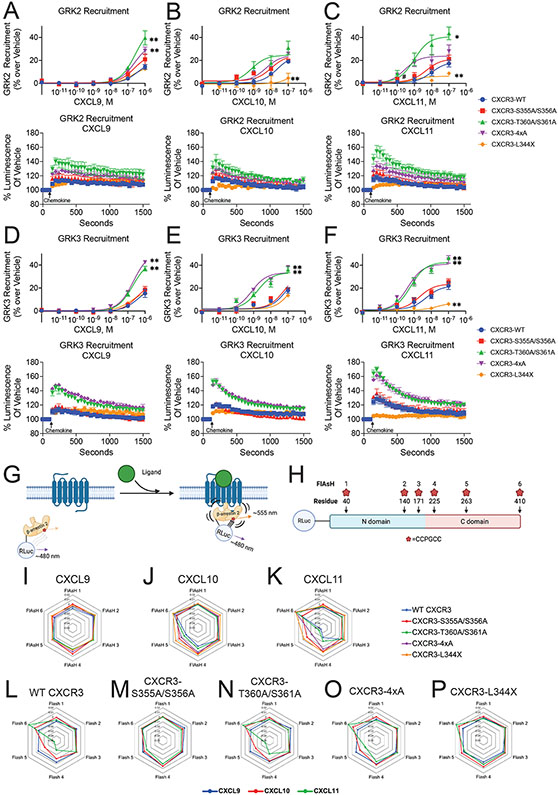
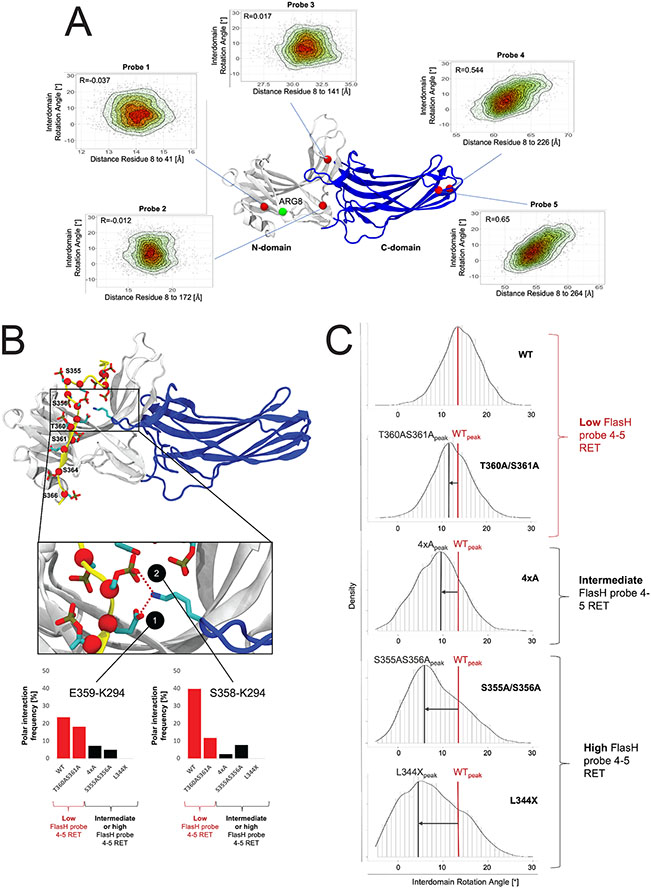
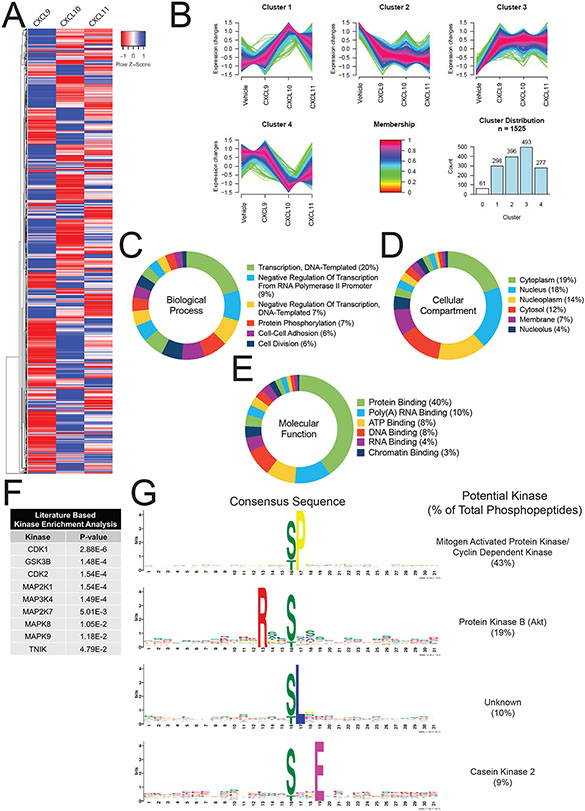
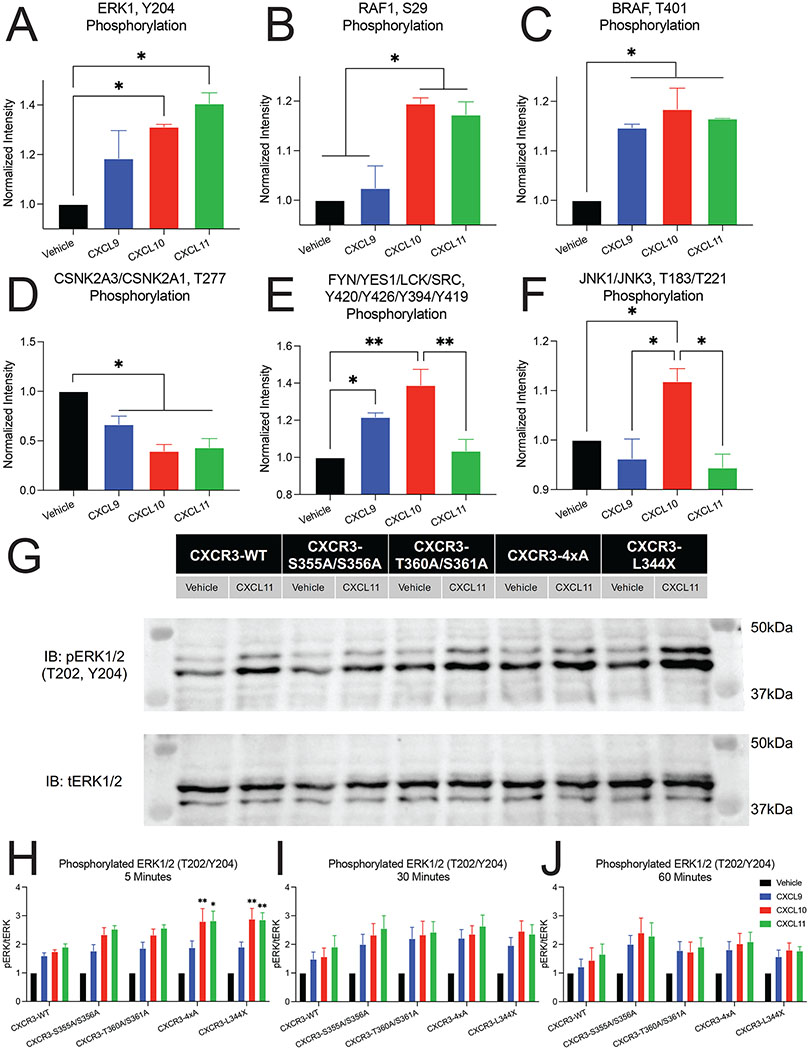
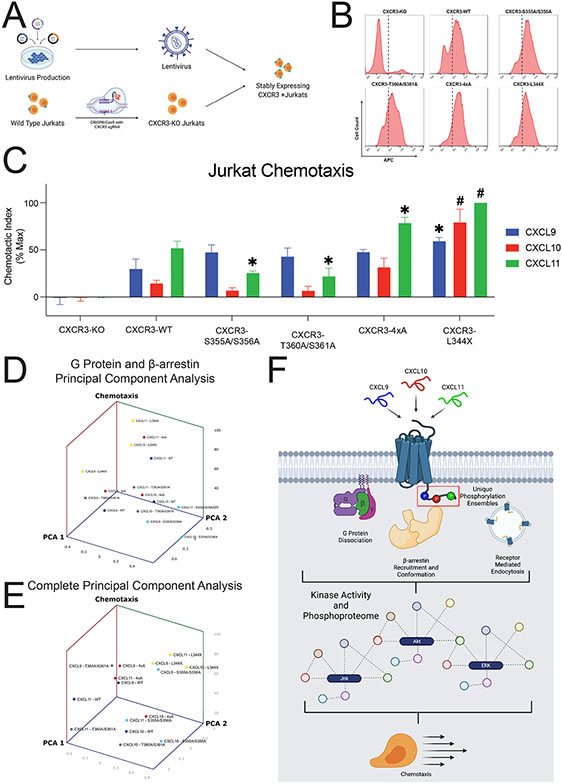
G protein-coupled receptor (GPCR)-biased agonism, selective activation of certain signaling pathways relative to others, is thought to be directed by differential GPCR phosphorylation "barcodes." At chemokine receptors, endogenous chemokines can act as "biased agonists", which may contribute to the limited success when pharmacologically targeting these receptors. Here, mass spectrometry-based global phosphoproteomics revealed that CXCR3 chemokines generate different phosphorylation barcodes associated with differential transducer activation. Chemokine stimulation resulted in distinct changes throughout the kinome in global phosphoproteomics studies. Mutation of CXCR3 phosphosites altered β-arrestin 2 conformation in cellular assays and was consistent with conformational changes observed in molecular dynamics simulations. T cells expressing phosphorylation-deficient CXCR3 mutants resulted in agonist- and receptor-specific chemotactic profiles. Our results demonstrate that CXCR3 chemokines are non-redundant and act as biased agonists through differential encoding of phosphorylation barcodes, leading to distinct physiological processes.
Keywords: CXCR3; G protein-coupled receptor; MAP kinase; biased agonism; chemokine; chemotaxis; mass spectrometry; molecular dynamics; phosphoproteomics; β-arrestin.
Copyright © 2023 Elsevier Ltd. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures
Update of
-
Phosphorylation barcodes direct biased chemokine signaling at CXCR3.bioRxiv [Preprint]. 2023 Mar 14:2023.03.14.532634. doi: 10.1101/2023.03.14.532634. bioRxiv. 2023. Update in: Cell Chem Biol. 2023 Apr 20;30(4):362-382.e8. doi: 10.1016/j.chembiol.2023.03.006. PMID: 36993369 Free PMC article. Updated. Preprint.
References
-
- Nobles KN, Xiao K, Ahn S, Shukla AK, Lam CM, Rajagopal S, Strachan RT, Huang TY, Bressler EA, Hara MR, et al. (2011). Distinct phosphorylation sites on the beta(2)-adrenergic receptor establish a barcode that encodes differential functions of beta-arrestin. Sci Signal 4, ra51. 10.1126/scisignal.2001707. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
