

The evolution and international spread of extensively drug resistant Shigella sonnei
- PMID: 37031199
- PMCID: PMC10082799
- DOI: 10.1038/s41467-023-37672-w
The evolution and international spread of extensively drug resistant Shigella sonnei
Erratum in
-
Author Correction: The evolution and international spread of extensively drug resistant Shigella sonnei.Nat Commun. 2023 Apr 21;14(1):2302. doi: 10.1038/s41467-023-38041-3. Nat Commun. 2023. PMID: 37085572 Free PMC article. No abstract available.
Abstract
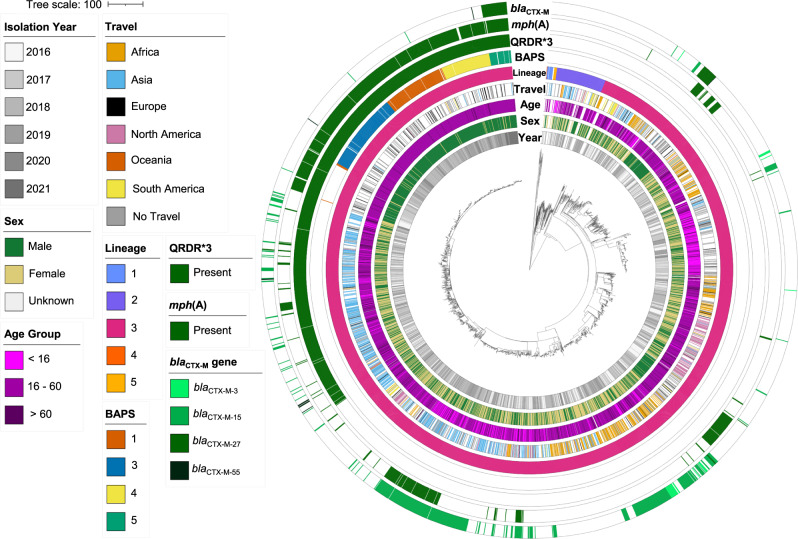
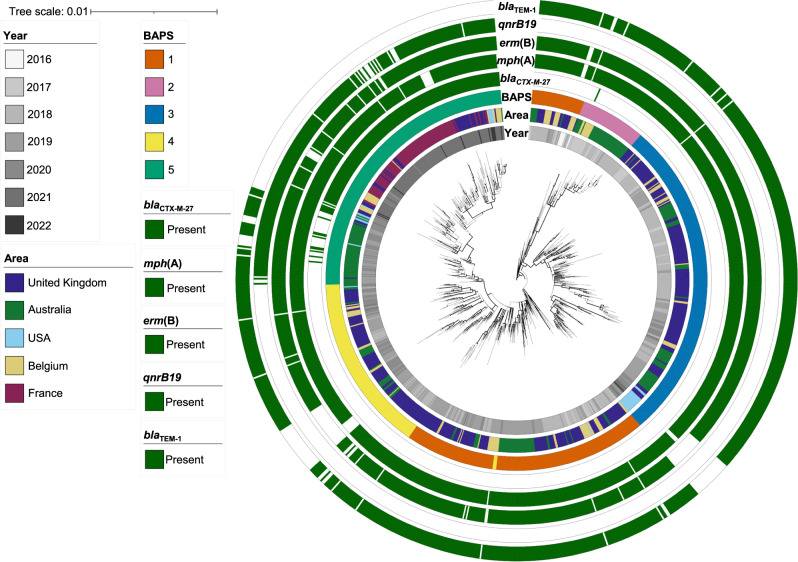
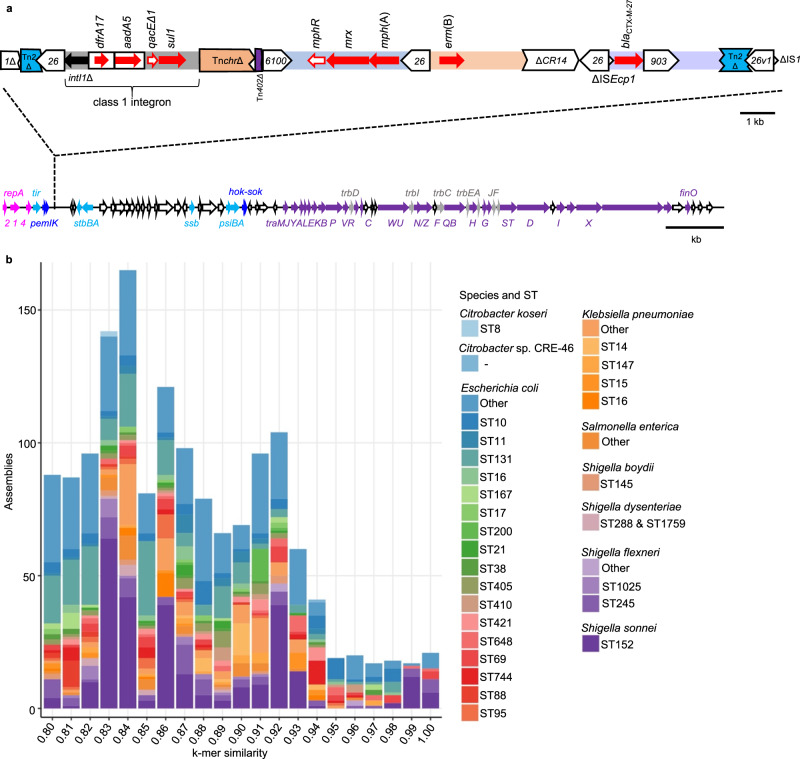
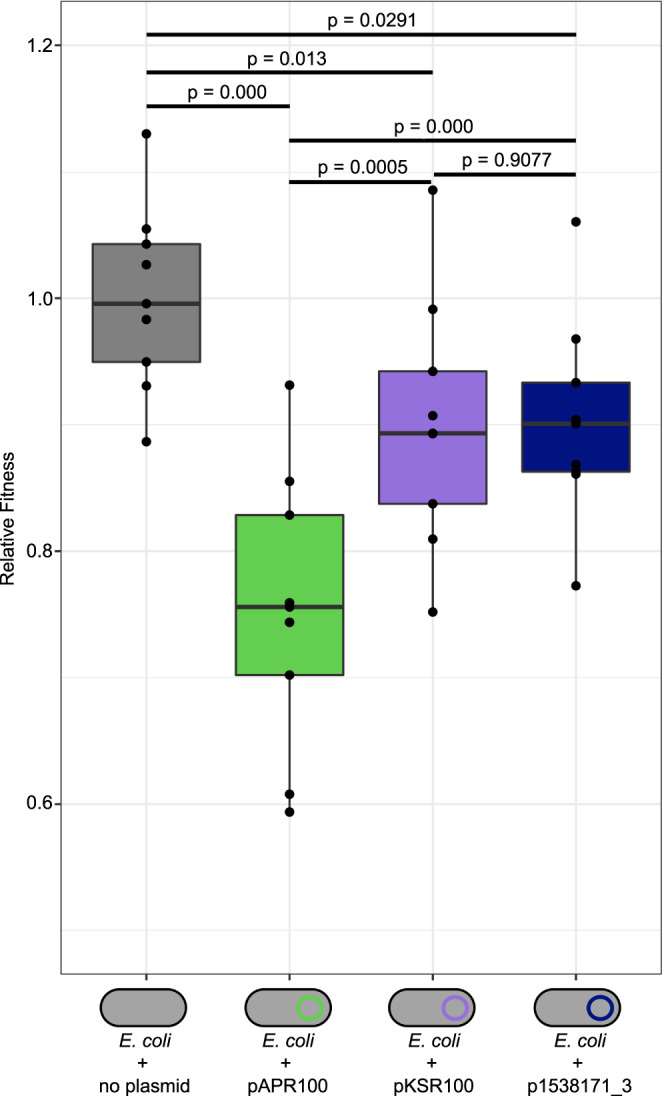
Shigella sonnei causes shigellosis, a severe gastrointestinal illness that is sexually transmissible among men who have sex with men (MSM). Multidrug resistance in S. sonnei is common including against World Health Organisation recommended treatment options, azithromycin, and ciprofloxacin. Recently, an MSM-associated outbreak of extended-spectrum β-lactamase producing, extensively drug resistant S. sonnei was reported in the United Kingdom. Here, we aimed to identify the genetic basis, evolutionary history, and international dissemination of the outbreak strain. Our genomic epidemiological analyses of 3,304 isolates from the United Kingdom, Australia, Belgium, France, and the United States of America revealed an internationally connected outbreak with a most recent common ancestor in 2018 carrying a low-fitness cost resistance plasmid, previously observed in travel associated sublineages of S. flexneri. Our results highlight the persistent threat of horizontally transmitted antimicrobial resistance and the value of continuing to work towards early and open international sharing of genomic surveillance data.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
