

This is a preprint.
Transcription-replication interactions reveal principles of bacterial genome regulation
- PMID: 37034646
- PMCID: PMC10081379
- DOI: 10.21203/rs.3.rs-2724389/v1
Transcription-replication interactions reveal principles of bacterial genome regulation
Update in
-
Transcription-replication interactions reveal bacterial genome regulation.Nature. 2024 Feb;626(7999):661-669. doi: 10.1038/s41586-023-06974-w. Epub 2024 Jan 24. Nature. 2024. PMID: 38267581 Free PMC article.
Abstract
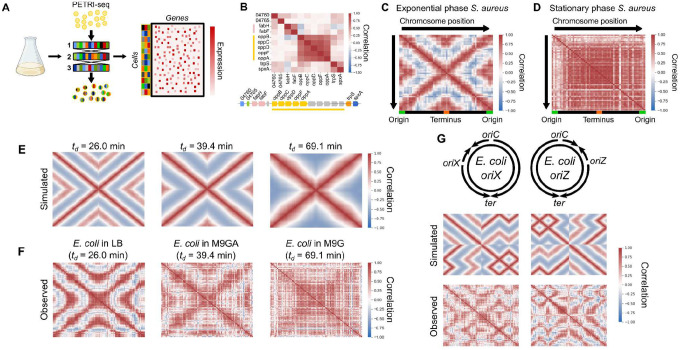
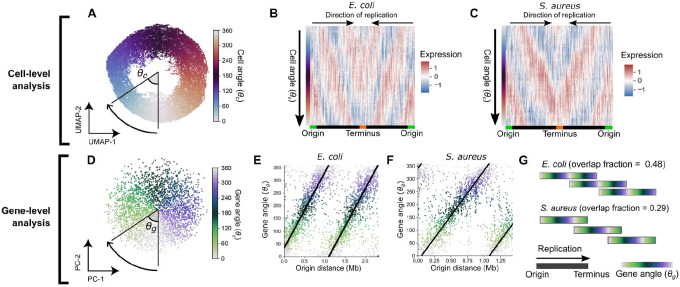
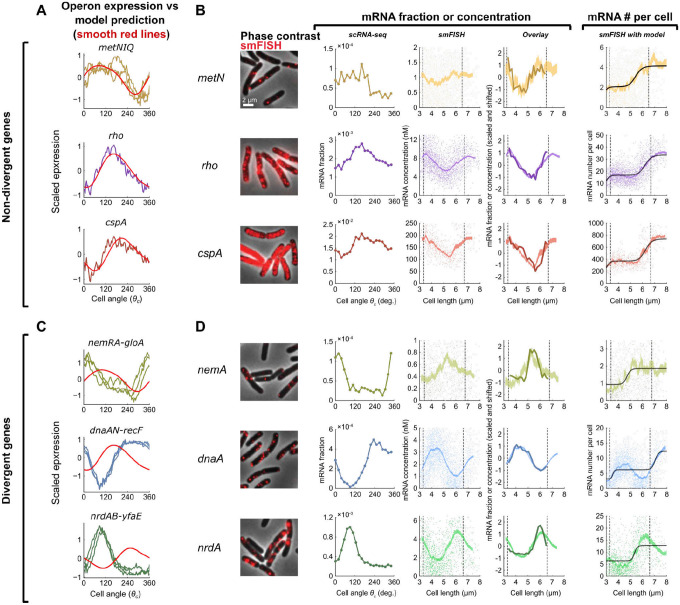
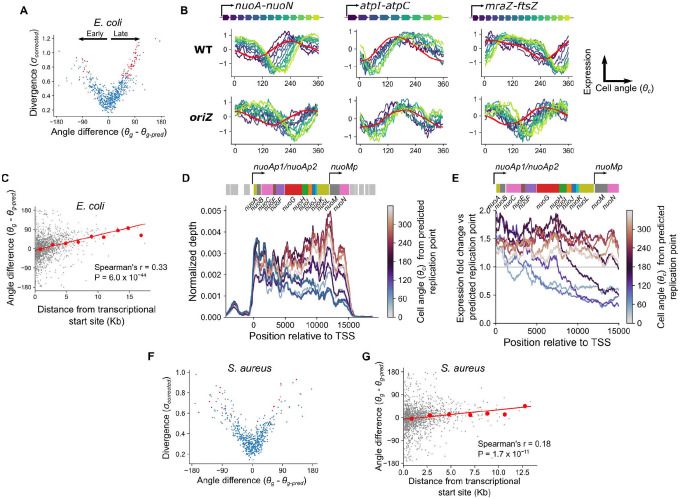
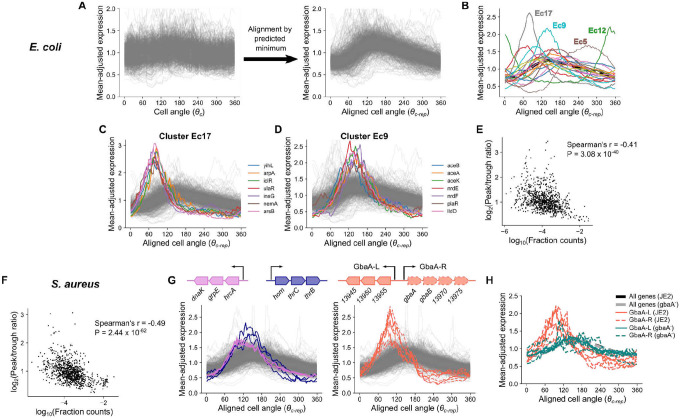
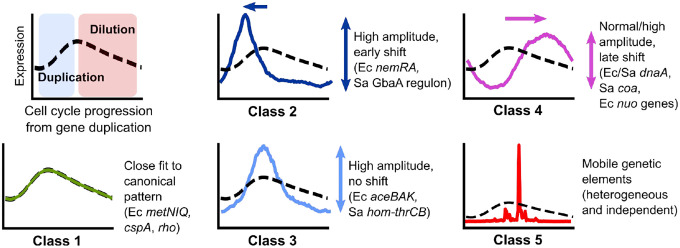
Organisms determine the transcription rates of thousands of genes through a few modes of regulation that recur across the genome1. These modes interact with a changing cellular environment to yield highly dynamic expression patterns2. In bacteria, the relationship between a gene's regulatory architecture and its expression is well understood for individual model gene circuits3,4. However, a broader perspective of these dynamics at the genome-scale is lacking, in part because bacterial transcriptomics have hitherto captured only a static snapshot of expression averaged across millions of cells5. As a result, the full diversity of gene expression dynamics and their relation to regulatory architecture remains unknown. Here we present a novel genome-wide classification of regulatory modes based on each gene's transcriptional response to its own replication, which we term the Transcription-Replication Interaction Profile (TRIP). We found that the response to the universal perturbation of chromosomal replication integrates biological regulatory factors with biophysical molecular events on the chromosome to reveal a gene's local regulatory context. While the TRIPs of many genes conform to a gene dosage-dependent pattern, others diverge in distinct ways, including altered timing or amplitude of expression, and this is shaped by factors such as intra-operon position, repression state, or presence on mobile genetic elements. Our transcriptome analysis also simultaneously captures global properties, such as the rates of replication and transcription, as well as the nestedness of replication patterns. This work challenges previous notions of the drivers of expression heterogeneity within a population of cells, and unearths a previously unseen world of gene transcription dynamics.
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures
References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
