

This is a preprint.
Proteostasis and lysosomal repair deficits in transdifferentiated neurons of Alzheimer's disease
- PMID: 37034684
- PMCID: PMC10081252
- DOI: 10.1101/2023.03.27.534444
Proteostasis and lysosomal repair deficits in transdifferentiated neurons of Alzheimer's disease
Update in
-
Proteostasis and lysosomal repair deficits in transdifferentiated neurons of Alzheimer's disease.Nat Cell Biol. 2025 Apr;27(4):619-632. doi: 10.1038/s41556-025-01623-y. Epub 2025 Mar 26. Nat Cell Biol. 2025. PMID: 40140603 Free PMC article.
Abstract
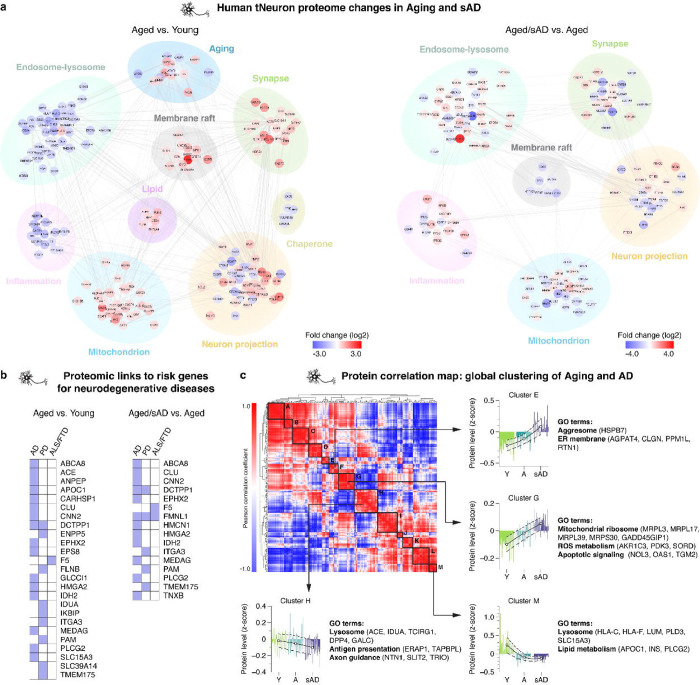
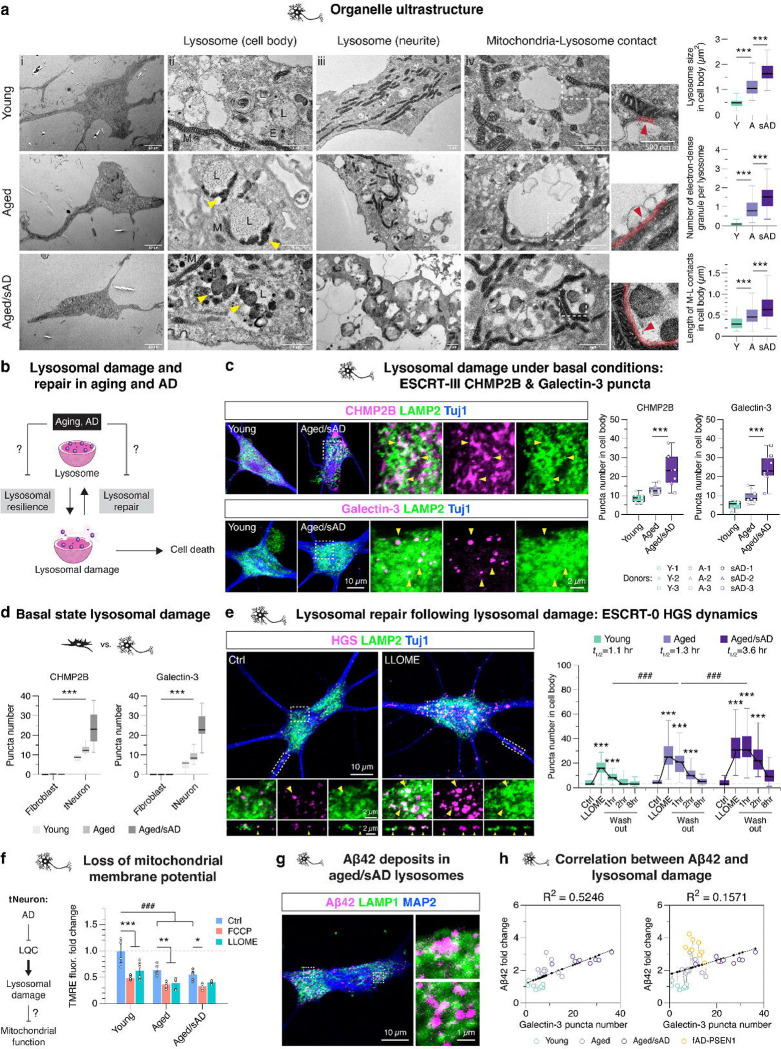
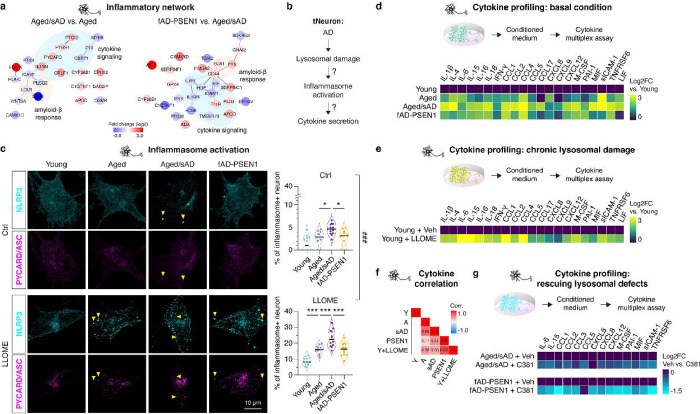
Aging is the most prominent risk factor for Alzheimer's disease (AD). However, the cellular mechanisms linking neuronal proteostasis decline to the characteristic aberrant protein deposits in AD brains remain elusive. Here, we develop transdifferentiated neurons (tNeurons) from human dermal fibroblasts as a neuronal model that retains aging hallmarks and exhibits AD-linked vulnerabilities. Remarkably, AD tNeurons accumulate proteotoxic deposits, including phospho-Tau and Aβ, resembling those in AD patient and APP mouse brains. Quantitative tNeuron proteomics identify aging and AD-linked deficits in proteostasis and organelle homeostasis, most notably in endosome-lysosomal components. Lysosomal deficits in aged tNeurons, including constitutive lysosomal damage and ESCRT-mediated lysosomal repair defects, are exacerbated in AD tNeurons and linked to inflammatory cytokine secretion and cell death. Supporting lysosomal deficits' centrality in AD, compounds ameliorating lysosomal function reduce Aβ deposits and cytokine secretion. Thus, the tNeuron model system reveals impaired lysosomal homeostasis as an early event of aging and AD.
Conflict of interest statement
Competing interests: R.V., J.L. and T.W.-C. are co-founders of Qinotto Inc. Other authors declare no competing interests.
Figures



References
References (Main)
References (Methods)
-
- Rockenstein E., Mallory M., Mante M., Sisk A. & Masliaha E. Early formation of mature amyloid-beta protein deposits in a mutant APP transgenic model depends on levels of Abeta(1–42). Journal of neuroscience research 66, 573–582 (2001). - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources