

Performance efficient macromolecular mechanics via sub-nanometer shape based coarse graining
- PMID: 37037809
- PMCID: PMC10086035
- DOI: 10.1038/s41467-023-37801-5
Performance efficient macromolecular mechanics via sub-nanometer shape based coarse graining
Abstract
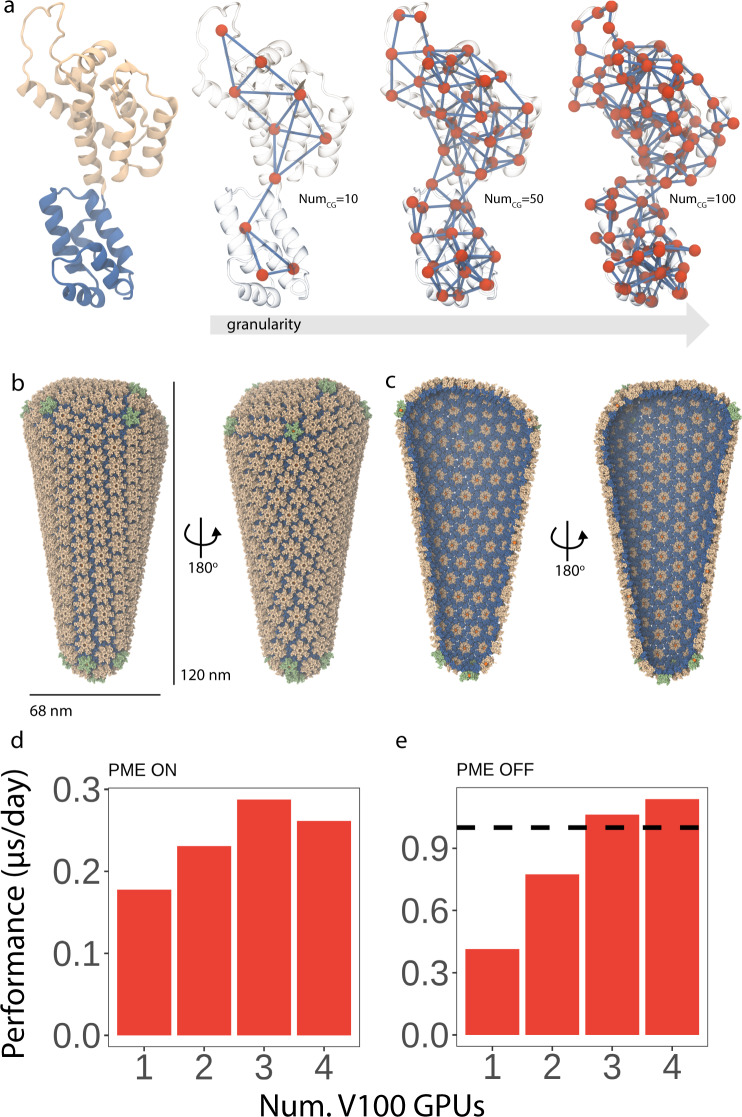
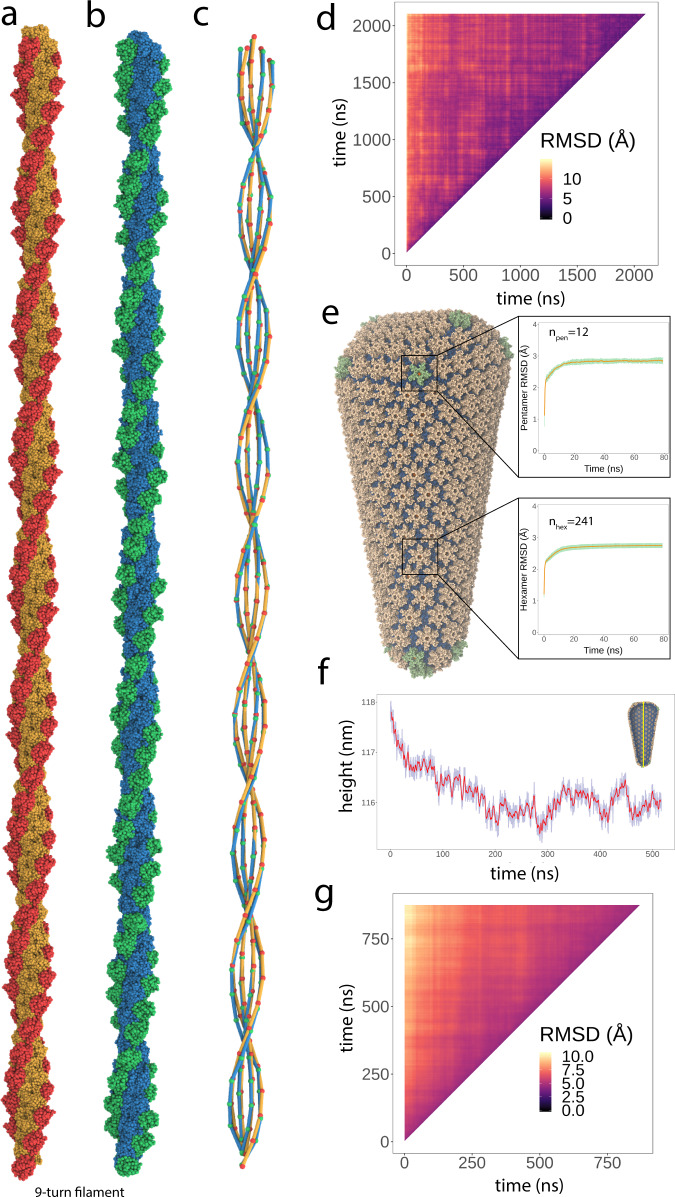
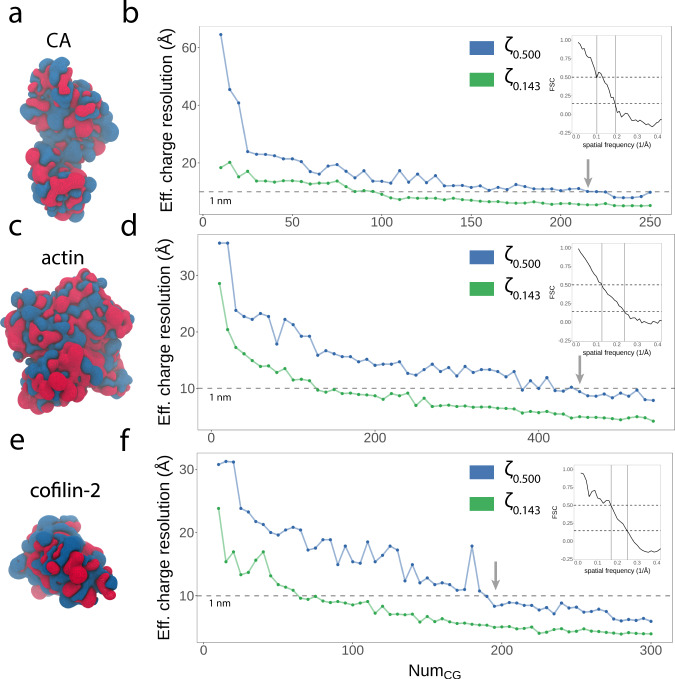
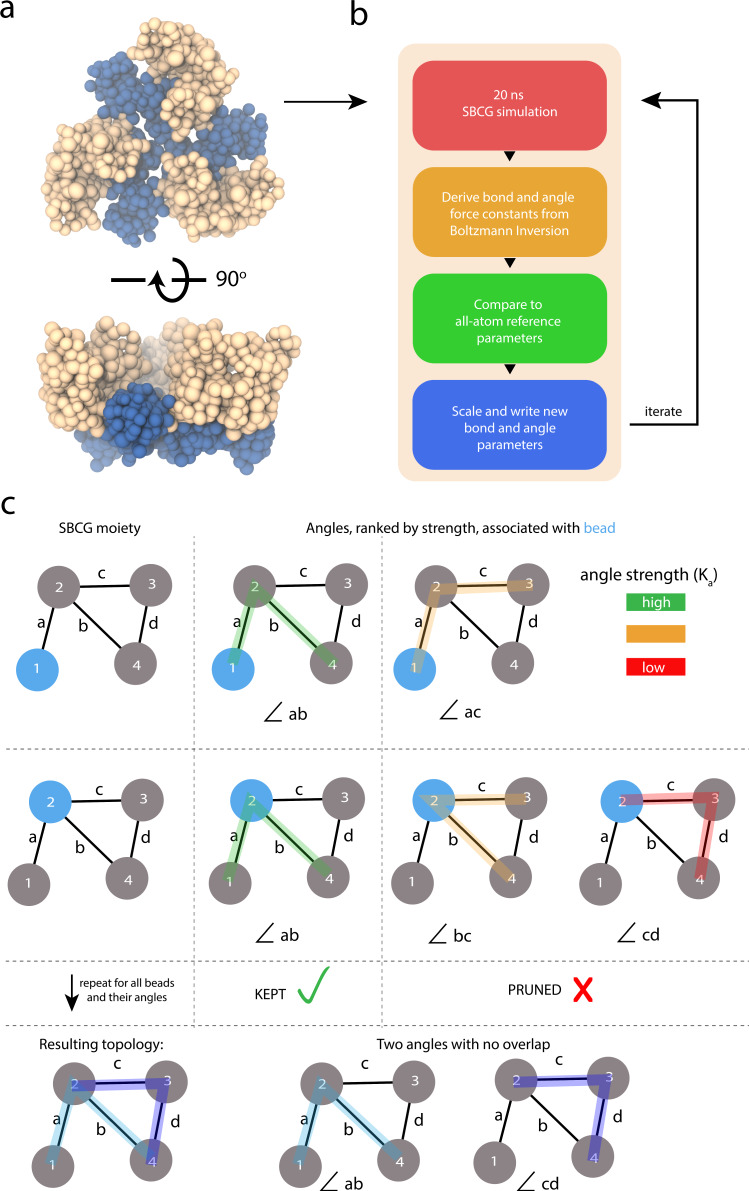
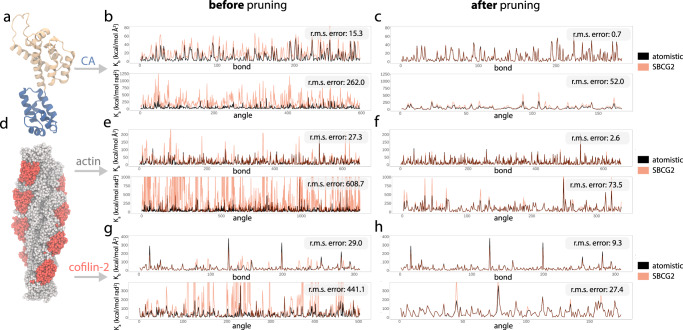
Dimensionality reduction via coarse grain modeling is a valuable tool in biomolecular research. For large assemblies, ultra coarse models are often knowledge-based, relying on a priori information to parameterize models thus hindering general predictive capability. Here, we present substantial advances to the shape based coarse graining (SBCG) method, which we refer to as SBCG2. SBCG2 utilizes a revitalized formulation of the topology representing network which makes high-granularity modeling possible, preserving atomistic details that maintain assembly characteristics. Further, we present a method of granularity selection based on charge density Fourier Shell Correlation and have additionally developed a refinement method to optimize, adjust and validate high-granularity models. We demonstrate our approach with the conical HIV-1 capsid and heteromultimeric cofilin-2 bound actin filaments. Our approach is available in the Visual Molecular Dynamics (VMD) software suite, and employs a CHARMM-compatible Hamiltonian that enables high-performance simulation in the GPU-resident NAMD3 molecular dynamics engine.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures



References
-
- Shaw DE, et al. Anton, a special-purpose machine for molecular dynamics simulation. Commun. ACM. 2008;51:91–97. doi: 10.1145/1364782.1364802. - DOI
-
- Cramer, C. J. Essentials of Computational Chemistry: Theories and Models (John Wiley & Sons, 2013).
-
- Nielsen SO, Lopez CF, Srinivas G, Klein ML. Coarse grain models and the computer simulation of soft materials. J. Phys. Condens. Matter. 2004;16:R481. doi: 10.1088/0953-8984/16/15/R03. - DOI
Grants and funding
LinkOut - more resources
Full Text Sources
