

Expanding known viral diversity in the healthy infant gut
- PMID: 37037943
- PMCID: PMC10159846
- DOI: 10.1038/s41564-023-01345-7
Expanding known viral diversity in the healthy infant gut
Abstract
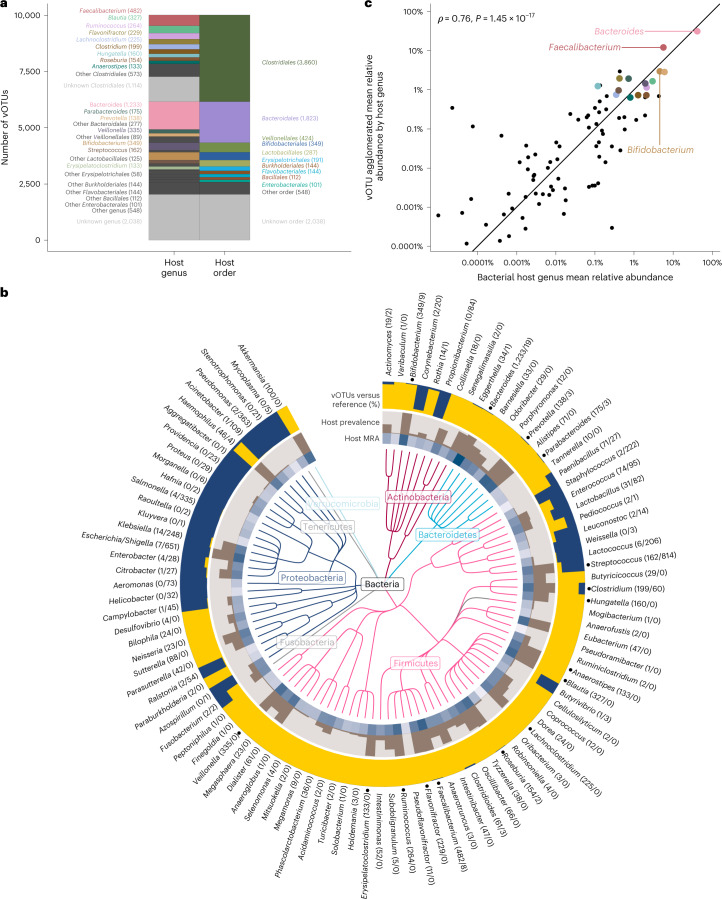
The gut microbiome is shaped through infancy and impacts the maturation of the immune system, thus protecting against chronic disease later in life. Phages, or viruses that infect bacteria, modulate bacterial growth by lysis and lysogeny, with the latter being especially prominent in the infant gut. Viral metagenomes (viromes) are difficult to analyse because they span uncharted viral diversity, lacking marker genes and standardized detection methods. Here we systematically resolved the viral diversity in faecal viromes from 647 1-year-olds belonging to Copenhagen Prospective Studies on Asthma in Childhood 2010, an unselected Danish cohort of healthy mother-child pairs. By assembly and curation we uncovered 10,000 viral species from 248 virus family-level clades (VFCs). Most (232 VFCs) were previously unknown, belonging to the Caudoviricetes viral class. Hosts were determined for 79% of phage using clustered regularly interspaced short palindromic repeat spacers within bacterial metagenomes from the same children. Typical Bacteroides-infecting crAssphages were outnumbered by undescribed phage families infecting Clostridiales and Bifidobacterium. Phage lifestyles were conserved at the viral family level, with 33 virulent and 118 temperate phage families. Virulent phages were more abundant, while temperate ones were more prevalent and diverse. Together, the viral families found in this study expand existing phage taxonomy and provide a resource aiding future infant gut virome research.
© 2023. The Author(s).
Conflict of interest statement
S.A.S. is a consultant for profluent.bio on a matter that is unrelated to the present study. D.S.N. has functioned as a consultant for the companies Pfizer and Sniprbiome on scientific matters not related to the present study. All remaining authors declare no conflicts of interest.
Figures












References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
