

Comparison of two multi-trait association testing methods and sequence-based fine mapping of six additive QTL in Swiss Large White pigs
- PMID: 37038103
- PMCID: PMC10084639
- DOI: 10.1186/s12864-023-09295-4
Comparison of two multi-trait association testing methods and sequence-based fine mapping of six additive QTL in Swiss Large White pigs
Abstract
Background: Genetic correlations between complex traits suggest that pleiotropic variants contribute to trait variation. Genome-wide association studies (GWAS) aim to uncover the genetic underpinnings of traits. Multivariate association testing and the meta-analysis of summary statistics from single-trait GWAS enable detecting variants associated with multiple phenotypes. In this study, we used array-derived genotypes and phenotypes for 24 reproduction, production, and conformation traits to explore differences between the two methods and used imputed sequence variant genotypes to fine-map six quantitative trait loci (QTL).
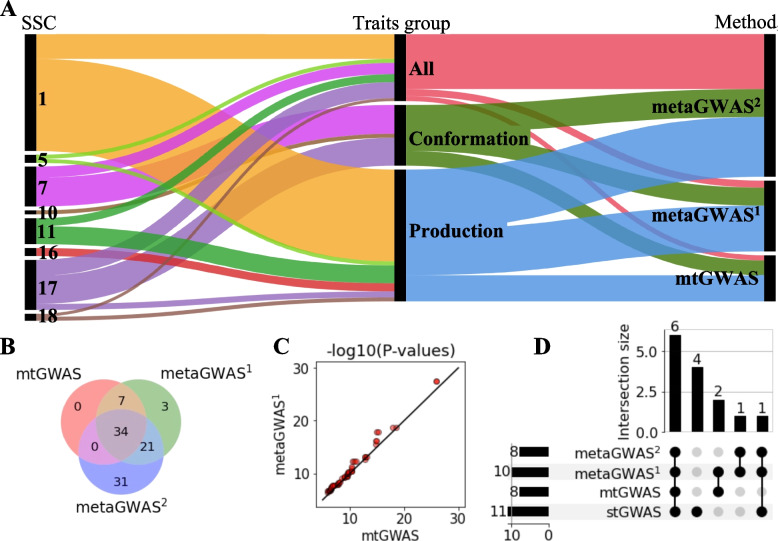
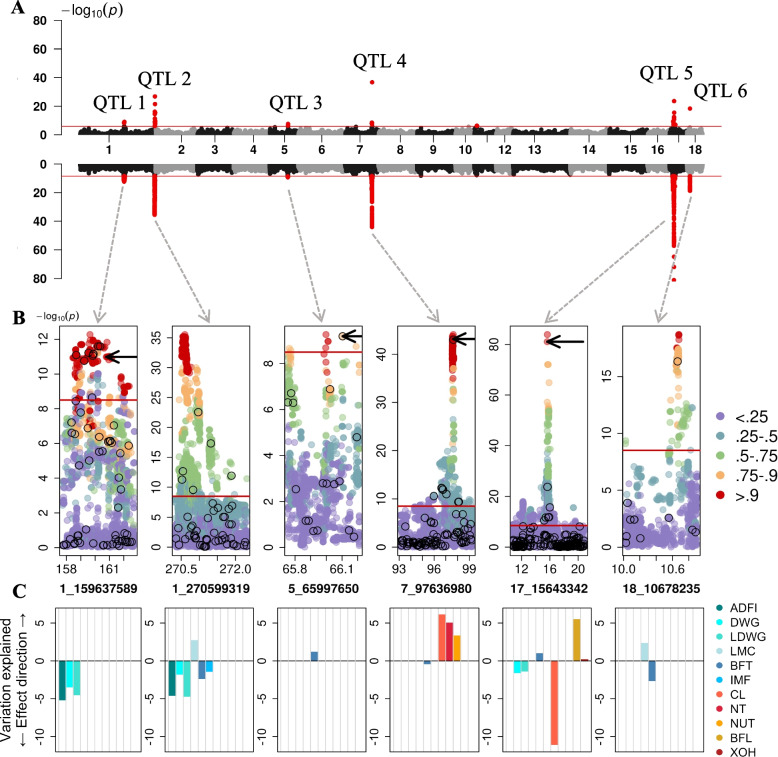
Results: We considered genotypes at 44,733 SNPs for 5,753 pigs from the Swiss Large White breed that had deregressed breeding values for 24 traits. Single-trait association analyses revealed eleven QTL that affected 15 traits. Multi-trait association testing and the meta-analysis of the single-trait GWAS revealed between 3 and 6 QTL, respectively, in three groups of traits. The multi-trait methods revealed three loci that were not detected in the single-trait GWAS. Four QTL that were identified in the single-trait GWAS, remained undetected in the multi-trait analyses. To pinpoint candidate causal variants for the QTL, we imputed the array-derived genotypes to the sequence level using a sequenced reference panel consisting of 421 pigs. This approach provided genotypes at 16 million imputed sequence variants with a mean accuracy of imputation of 0.94. The fine-mapping of six QTL with imputed sequence variant genotypes revealed four previously proposed causal mutations among the top variants.
Conclusions: Our findings in a medium-size cohort of pigs suggest that multivariate association testing and the meta-analysis of summary statistics from single-trait GWAS provide very similar results. Although multi-trait association methods provide a useful overview of pleiotropic loci segregating in mapping populations, the investigation of single-trait association studies is still advised, as multi-trait methods may miss QTL that are uncovered in single-trait GWAS.
Keywords: Genome-wide association study; Imputation; Meta-analyses; Multivariate analyses; Pleiotropy.
© 2023. The Author(s).
Conflict of interest statement
AH is employee of SUISAG (the Swiss pig breeding and competence centre). HP is a member of the editorial board of BMC Genomics. All other authors declare that they have no competing interests.
Figures
Similar articles
-
Multi-trait meta-analyses reveal 25 quantitative trait loci for economically important traits in Brown Swiss cattle.BMC Genomics. 2019 Sep 3;20(1):695. doi: 10.1186/s12864-019-6066-6. BMC Genomics. 2019. PMID: 31481029 Free PMC article.
-
Meta-analysis of sequence-based association studies across three cattle breeds reveals 25 QTL for fat and protein percentages in milk at nucleotide resolution.BMC Genomics. 2017 Nov 9;18(1):853. doi: 10.1186/s12864-017-4263-8. BMC Genomics. 2017. PMID: 29121857 Free PMC article.
-
A multi-trait meta-analysis with imputed sequence variants reveals twelve QTL for mammary gland morphology in Fleckvieh cattle.Genet Sel Evol. 2016 Feb 16;48:14. doi: 10.1186/s12711-016-0190-4. Genet Sel Evol. 2016. PMID: 26883850 Free PMC article.
-
Genetic Mapping of Quantitative Trait Loci for Egg Production and Egg Quality Traits in Chickens: a Review.J Poult Sci. 2017 Jan 25;54(1):1-12. doi: 10.2141/jpsa.0160121. J Poult Sci. 2017. PMID: 32908402 Free PMC article. Review.
-
'Particle genetics': treating every cell as unique.Trends Genet. 2014 Feb;30(2):49-56. doi: 10.1016/j.tig.2013.11.002. Epub 2013 Dec 6. Trends Genet. 2014. PMID: 24315431 Free PMC article. Review.
Cited by
-
Genome-Wide Association Study Meta-Analysis Elucidates Genetic Structure and Identifies Candidate Genes of Teat Number Traits in Pigs.Int J Mol Sci. 2023 Dec 29;25(1):451. doi: 10.3390/ijms25010451. Int J Mol Sci. 2023. PMID: 38203622 Free PMC article.
-
Unravelling novel and pleiotropic genes for cannon bone circumference and bone mineral density in Yorkshire pigs.J Anim Sci. 2024 Jan 3;102:skae036. doi: 10.1093/jas/skae036. J Anim Sci. 2024. PMID: 38330300 Free PMC article.
-
Single-variant genome-wide association study and regional heritability mapping of protein efficiency and performance traits in Large White pigs.Genet Sel Evol. 2025 Aug 14;57(1):45. doi: 10.1186/s12711-025-00993-z. Genet Sel Evol. 2025. PMID: 40813973 Free PMC article.
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
