

Computational Methods for Single-Cell Proteomics
- PMID: 37040735
- PMCID: PMC10621466
- DOI: 10.1146/annurev-biodatasci-020422-050255
Computational Methods for Single-Cell Proteomics
Abstract
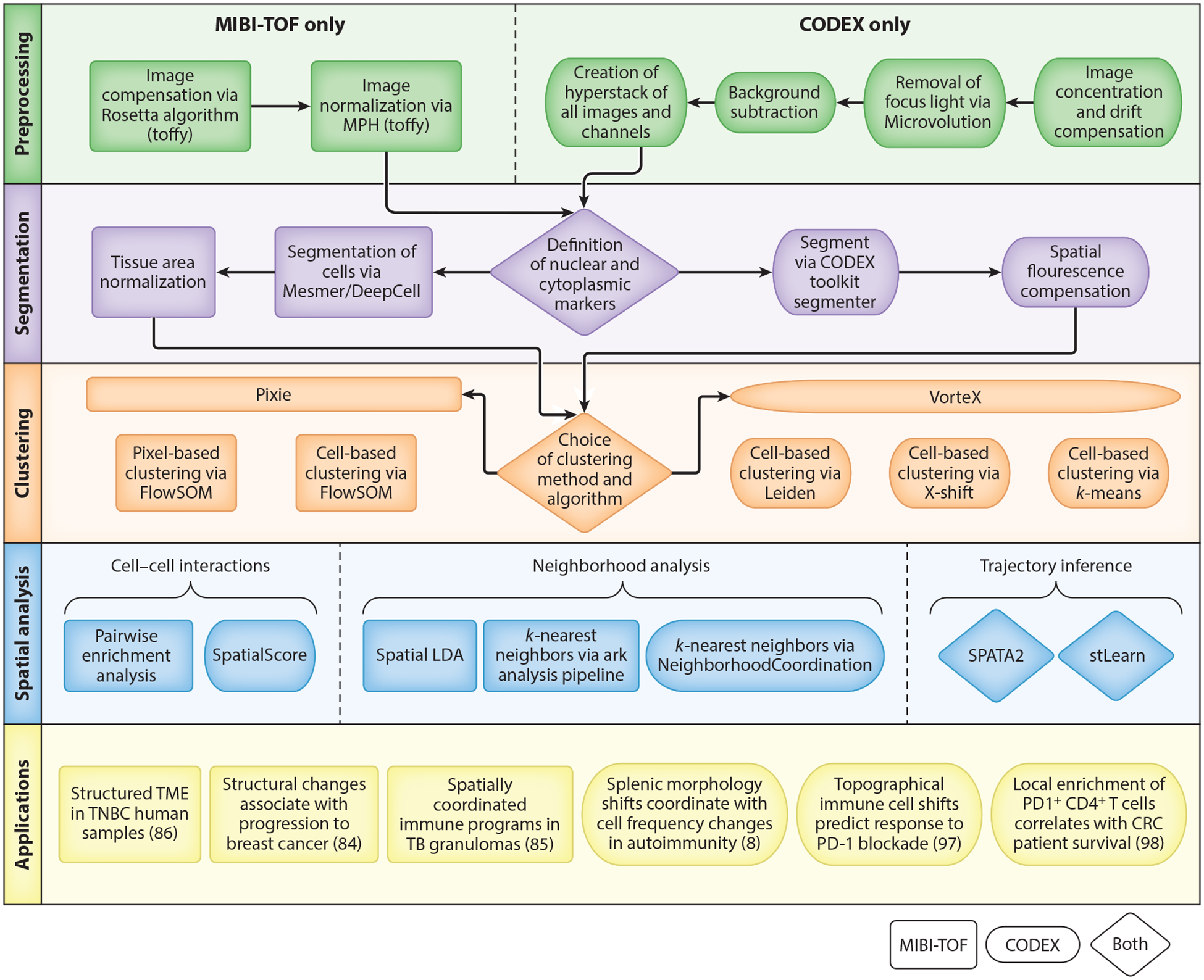
Advances in single-cell proteomics technologies have resulted in high-dimensional datasets comprising millions of cells that are capable of answering key questions about biology and disease. The advent of these technologies has prompted the development of computational tools to process and visualize the complex data. In this review, we outline the steps of single-cell and spatial proteomics analysis pipelines. In addition to describing available methods, we highlight benchmarking studies that have identified advantages and pitfalls of the currently available computational toolkits. As these technologies continue to advance, robust analysis tools should be developed in tandem to take full advantage of the potential biological insights provided by these data.
Keywords: clustering; computational methods; data analysis; mass cytometry; spatial proteomics; trajectory inference.
Figures
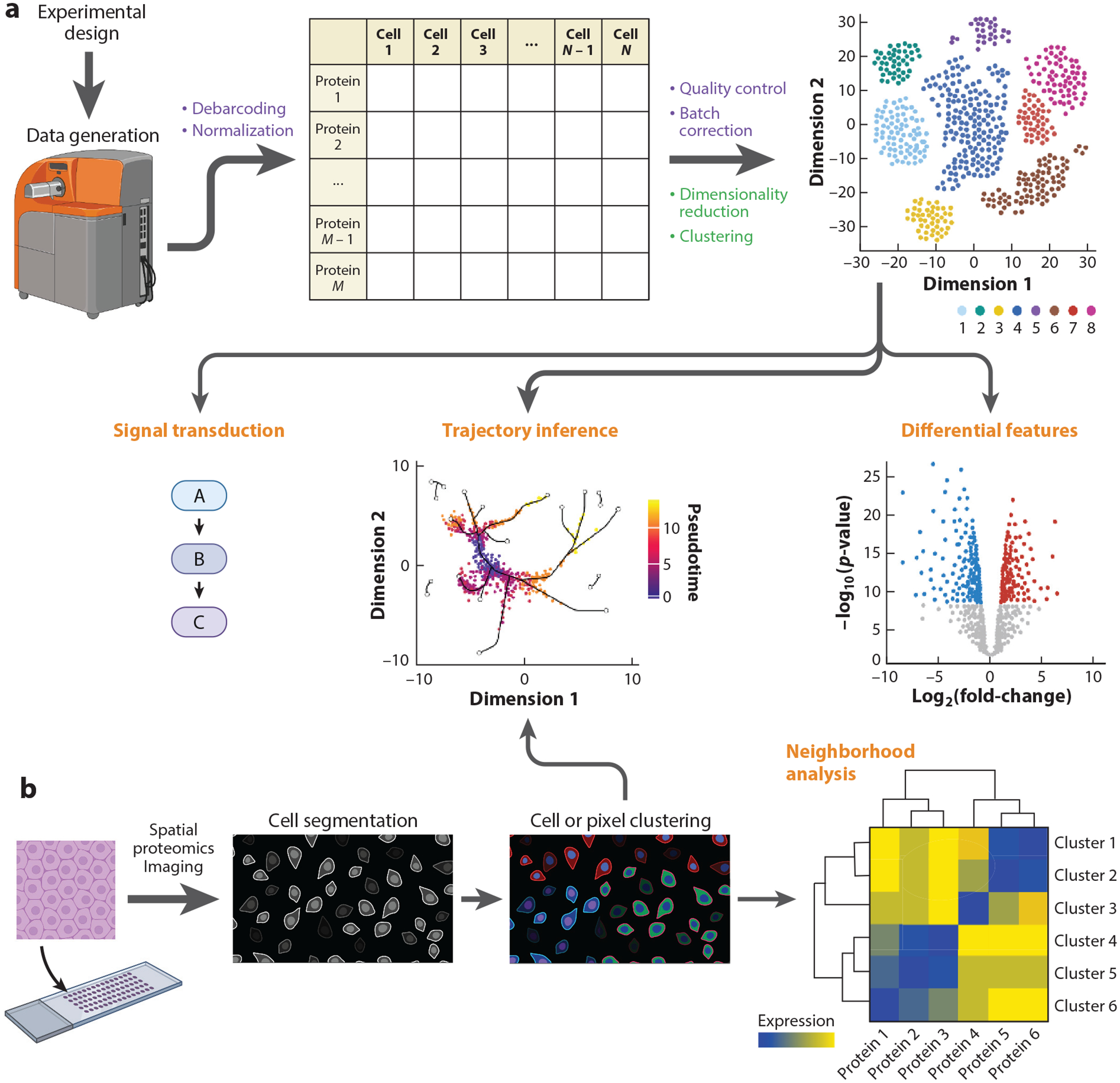
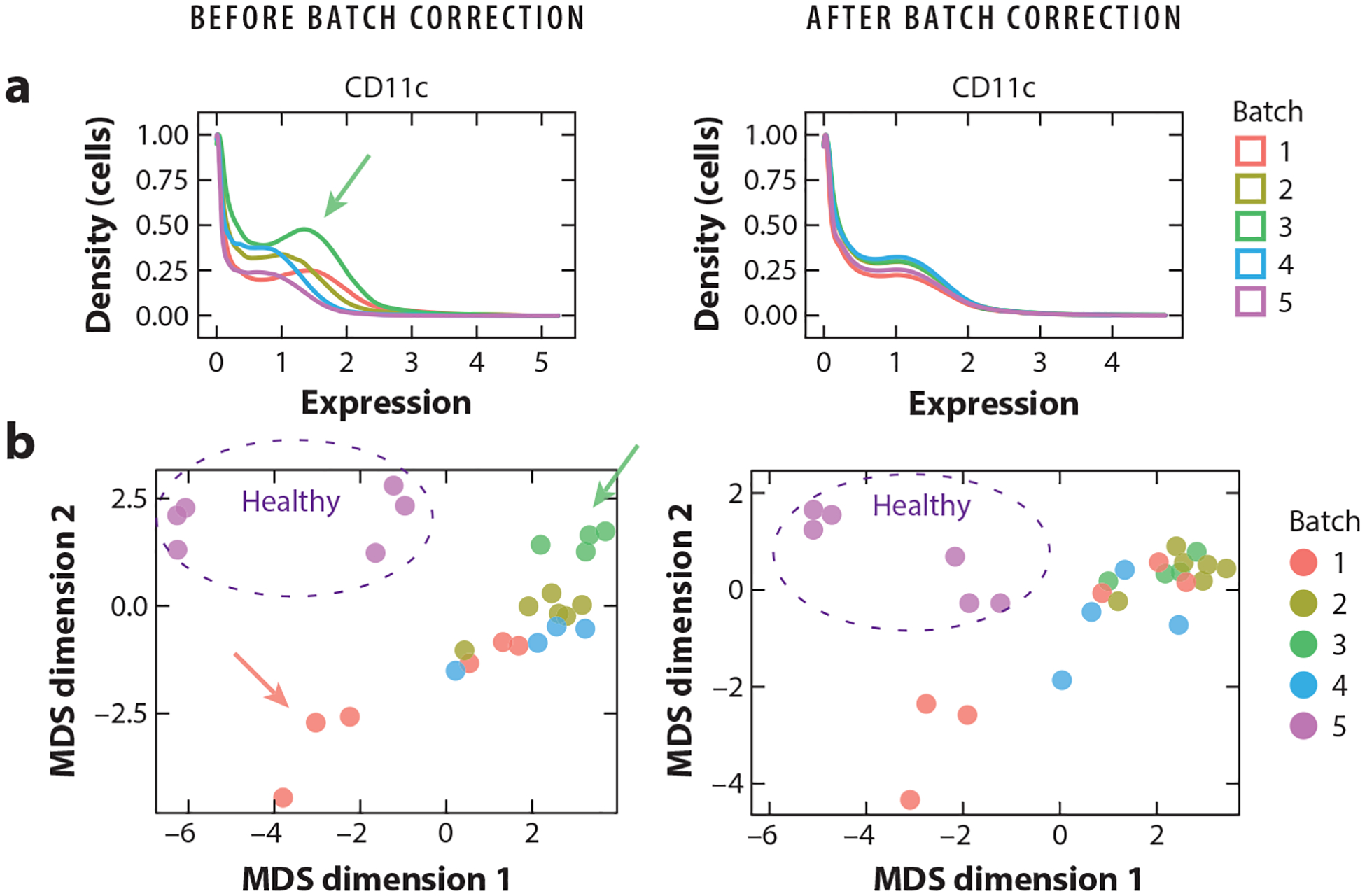
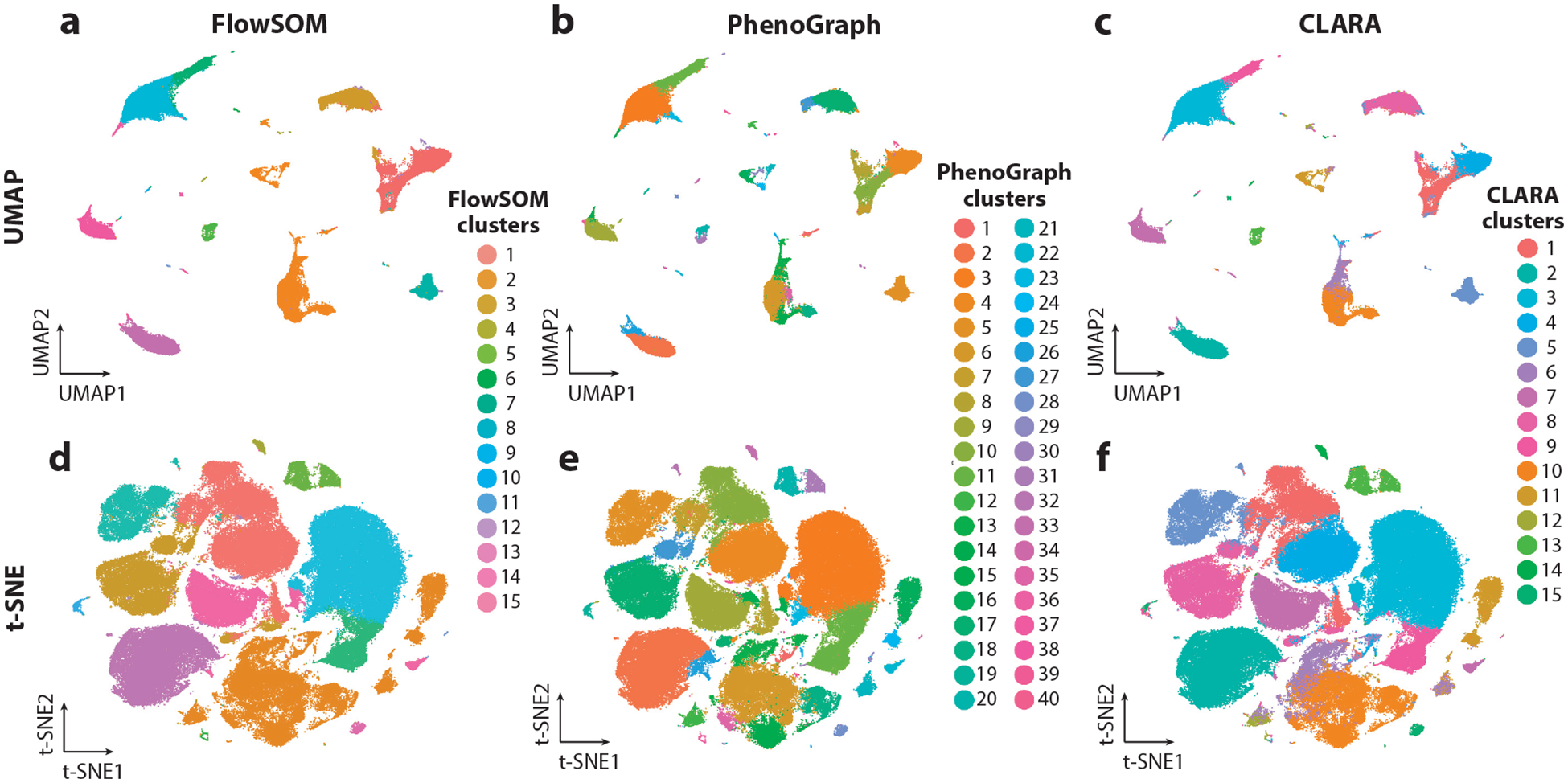
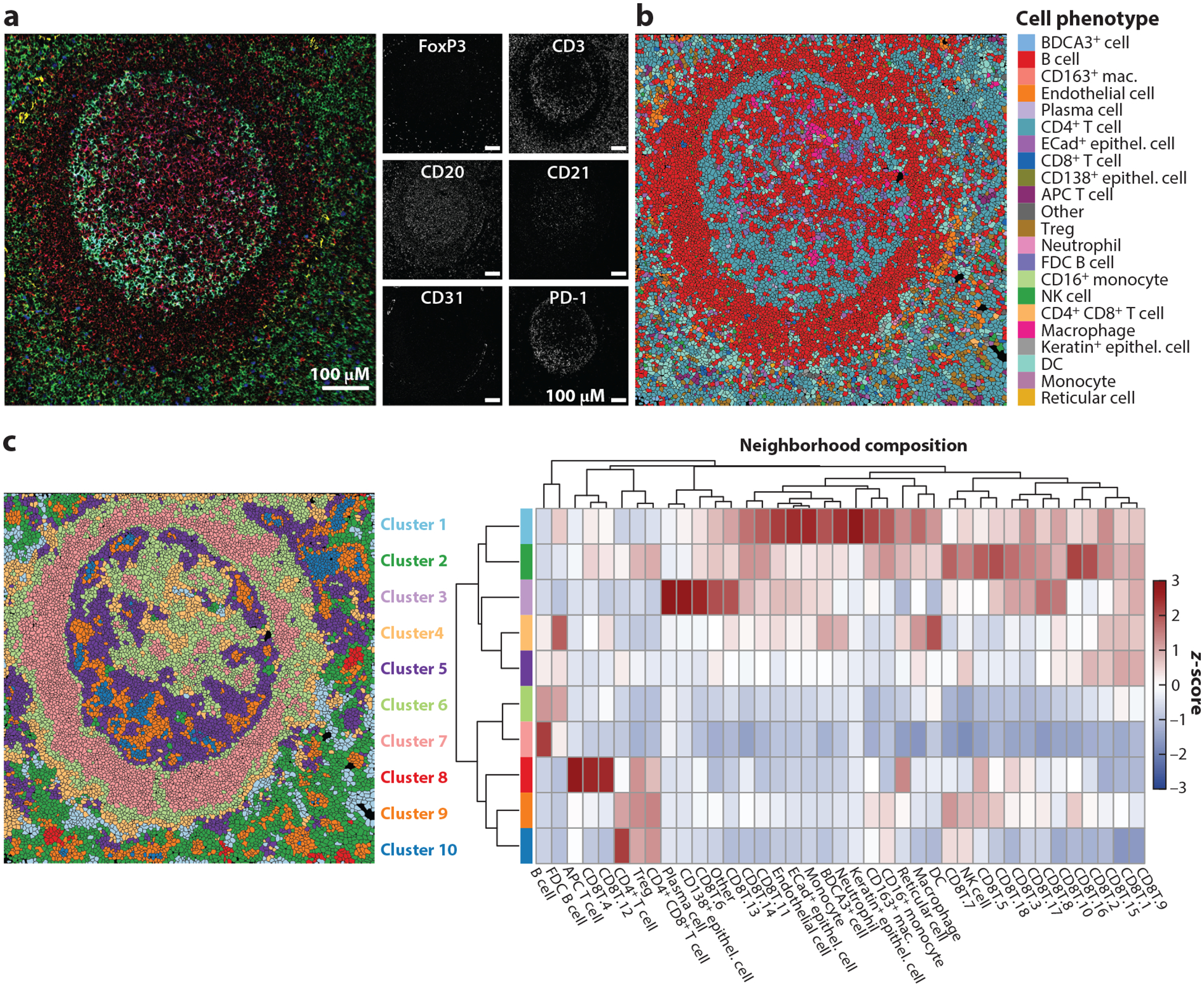
References
-
- Ornatsky OI, Lou X, Nitz M, Schäfer S, Sheldrick WS, et al. 2008. Study of cell antigens and intracellular DNA by identification of element-containing labels and metallointercalators using inductively coupled plasma mass spectrometry. Anal. Chem 80(7):2539–47 - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
