

FOXI3 pathogenic variants cause one form of craniofacial microsomia
- PMID: 37041148
- PMCID: PMC10090152
- DOI: 10.1038/s41467-023-37703-6
FOXI3 pathogenic variants cause one form of craniofacial microsomia
Abstract
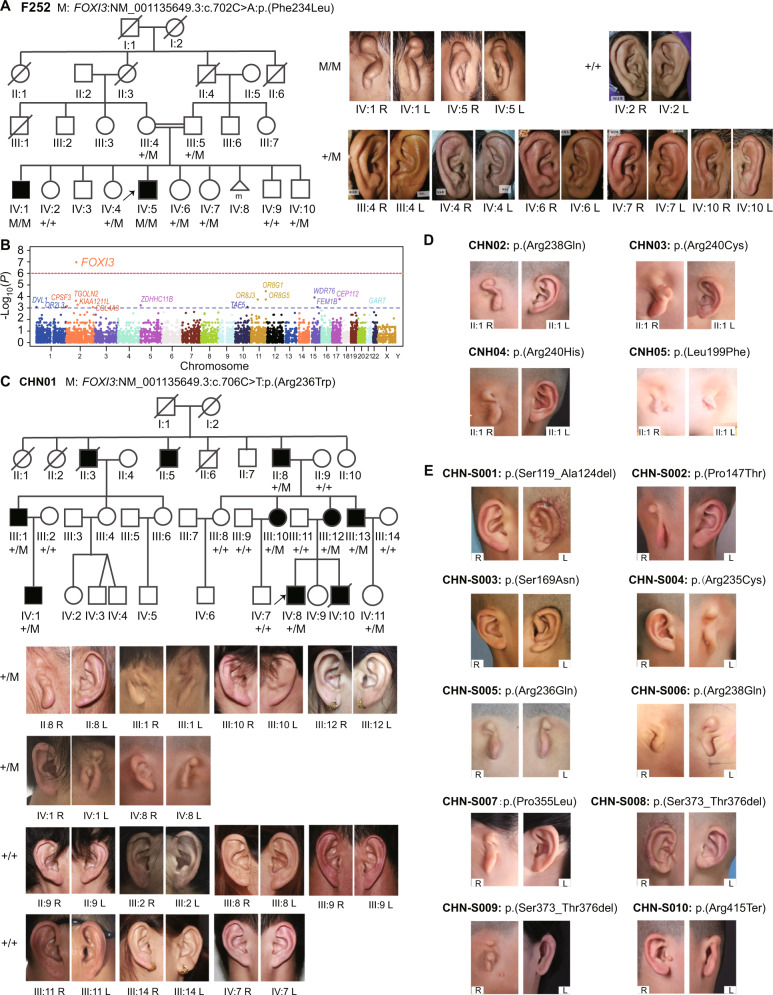
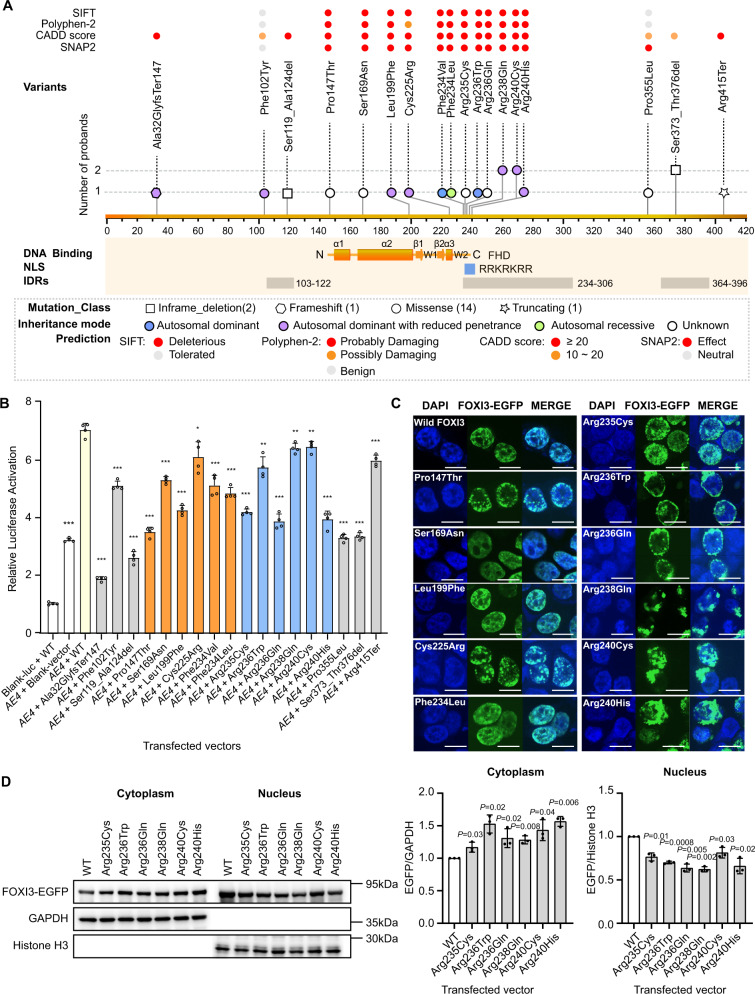
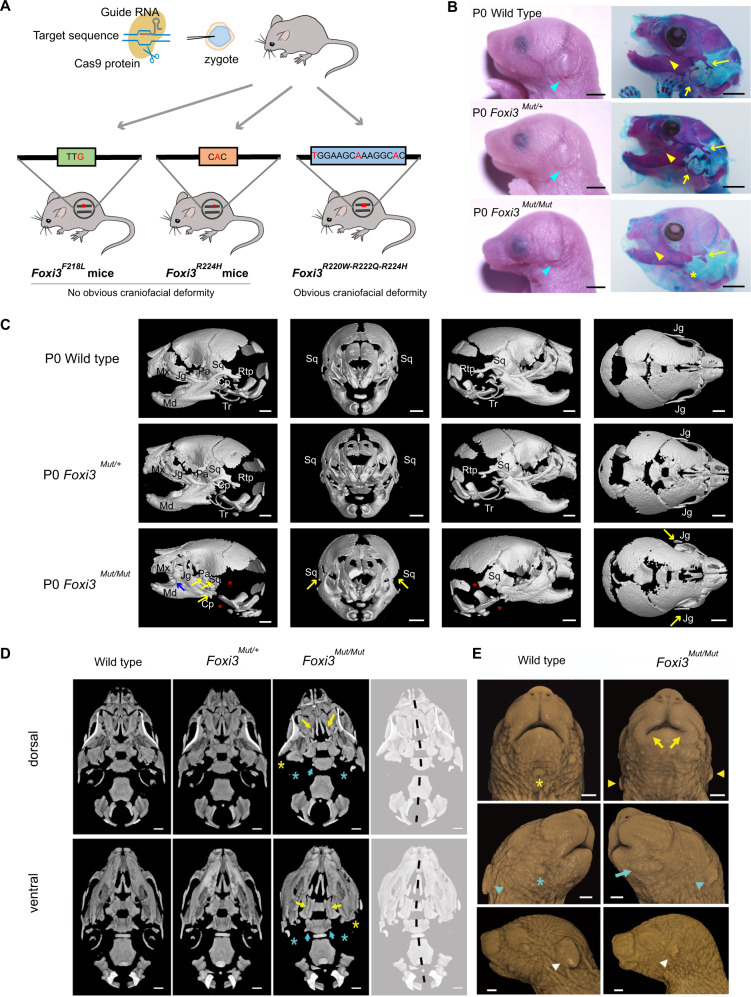
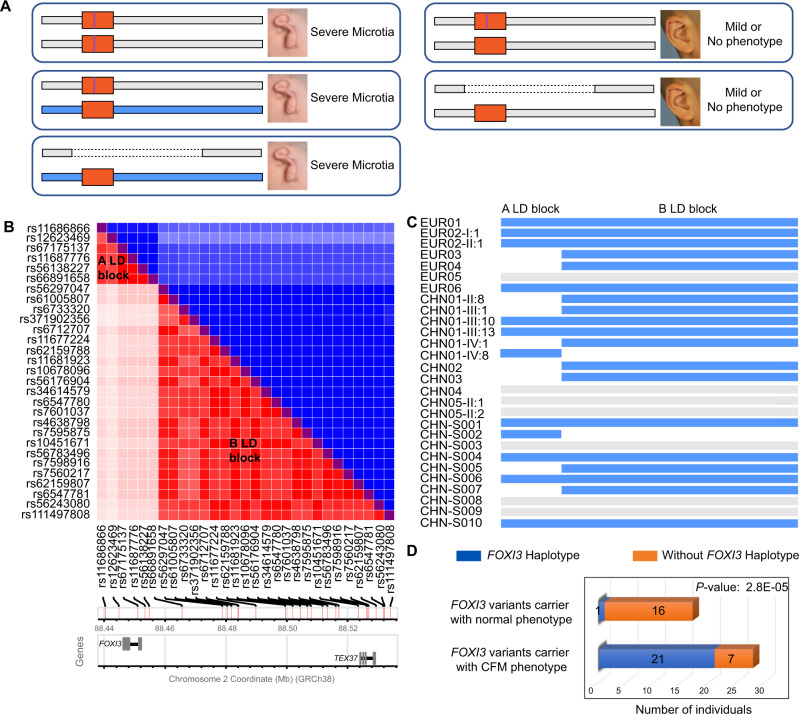
Craniofacial microsomia (CFM; also known as Goldenhar syndrome), is a craniofacial developmental disorder of variable expressivity and severity with a recognizable set of abnormalities. These birth defects are associated with structures derived from the first and second pharyngeal arches, can occur unilaterally and include ear dysplasia, microtia, preauricular tags and pits, facial asymmetry and other malformations. The inheritance pattern is controversial, and the molecular etiology of this syndrome is largely unknown. A total of 670 patients belonging to unrelated pedigrees with European and Chinese ancestry with CFM, are investigated. We identify 18 likely pathogenic variants in 21 probands (3.1%) in FOXI3. Biochemical experiments on transcriptional activity and subcellular localization of the likely pathogenic FOXI3 variants, and knock-in mouse studies strongly support the involvement of FOXI3 in CFM. Our findings indicate autosomal dominant inheritance with reduced penetrance, and/or autosomal recessive inheritance. The phenotypic expression of the FOXI3 variants is variable. The penetrance of the likely pathogenic variants in the seemingly dominant form is reduced, since a considerable number of such variants in affected individuals were inherited from non-affected parents. Here we provide suggestive evidence that common variation in the FOXI3 allele in trans with the pathogenic variant could modify the phenotypic severity and accounts for the incomplete penetrance.
© 2023. The Author(s).
Conflict of interest statement
S.E.A. is a cofounder and CEO of Medigenome, and serves on the Scientific Advisory Board of the “Imaging” institute in Paris; J.R.L. has stock ownership in 23andMe, is a paid consultant for Regeneron Genetics Center, is a co-inventor on multiple United States and European patents related to molecular diagnostics for inherited neuropathies, eye diseases, and bacterial genomic fingerprinting, and serves on the Scientific Advisory Board of BG. The Department of Molecular and Human Genetics at Baylor College of Medicine receives revenue from clinical genetic and genomic testing conducted at Baylor Genetics (BG) Laboratories. The remaining authors declare no competing interests.
Figures
References
-
- Goldenhar M. Associations malformatives de l’oeil et de l’oreille, en particulier le svndrome dermoide epibulbaire- appendices auriculaires-fistula auris congenita et ses relations avec la dysostose mandibulo-faciale. J. Genet. Hum. 1952;1:243–282.
-
- Cohen MM, Jr., Rollnick BR, Kaye CI. Oculoauriculovertebral spectrum: an updated critique. Cleft Palate J. 1989;26:276–286. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
