

A Diverse Virome Is Identified in Parasitic Flatworms of Domestic Animals in Xinjiang, China
- PMID: 37042768
- PMCID: PMC10269781
- DOI: 10.1128/spectrum.00702-23
A Diverse Virome Is Identified in Parasitic Flatworms of Domestic Animals in Xinjiang, China
Abstract
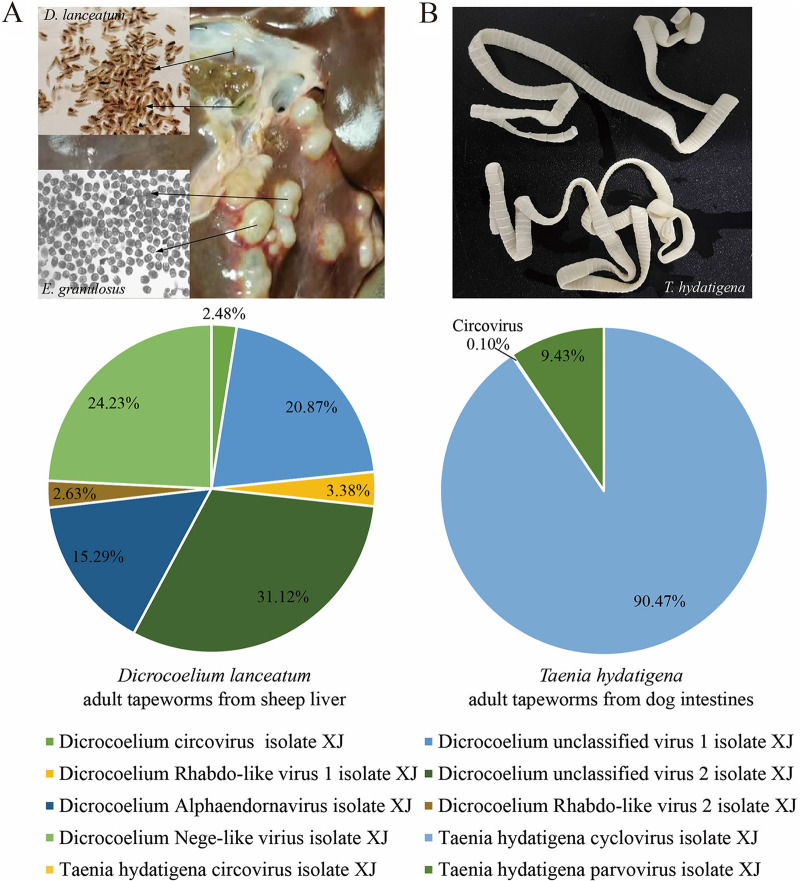
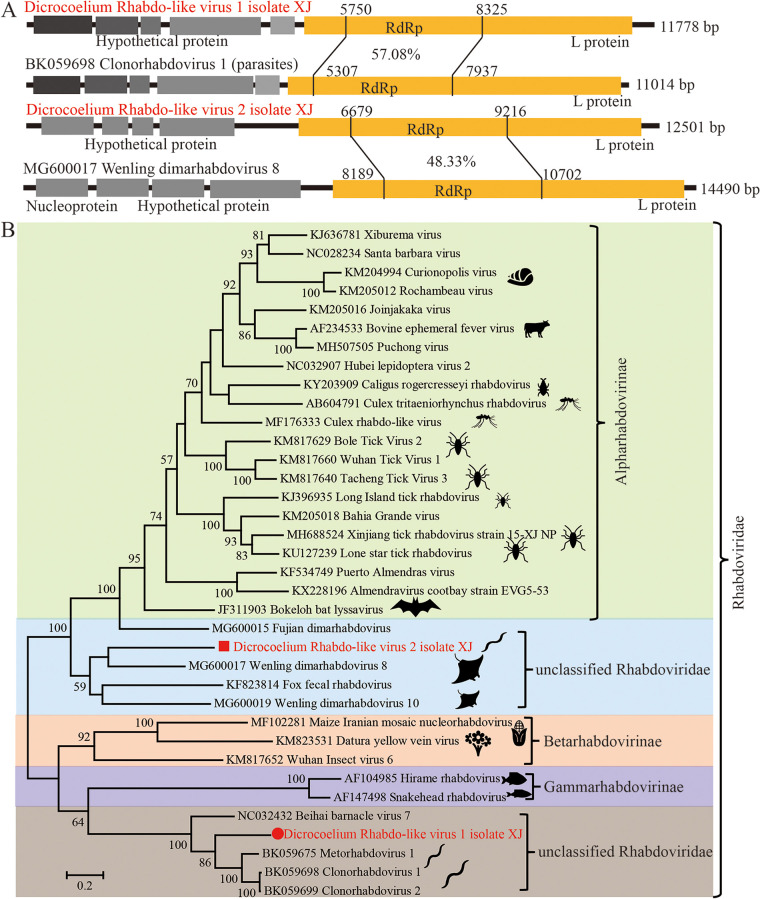
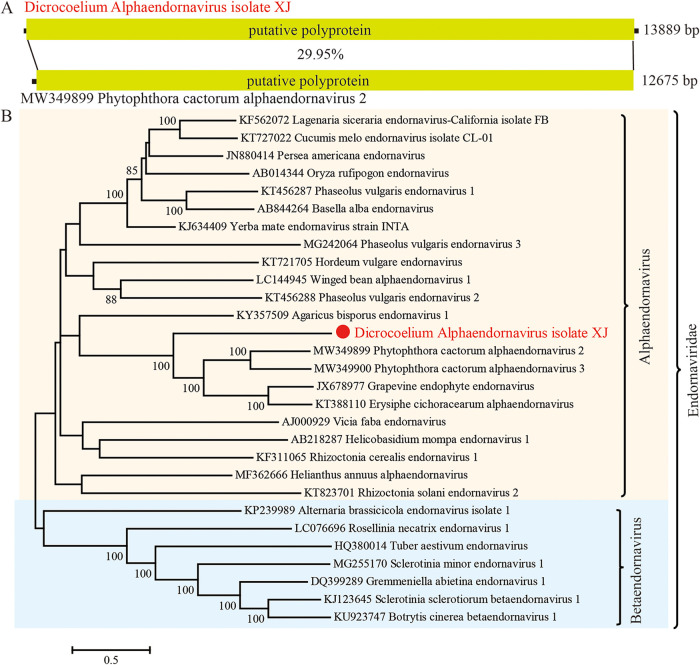
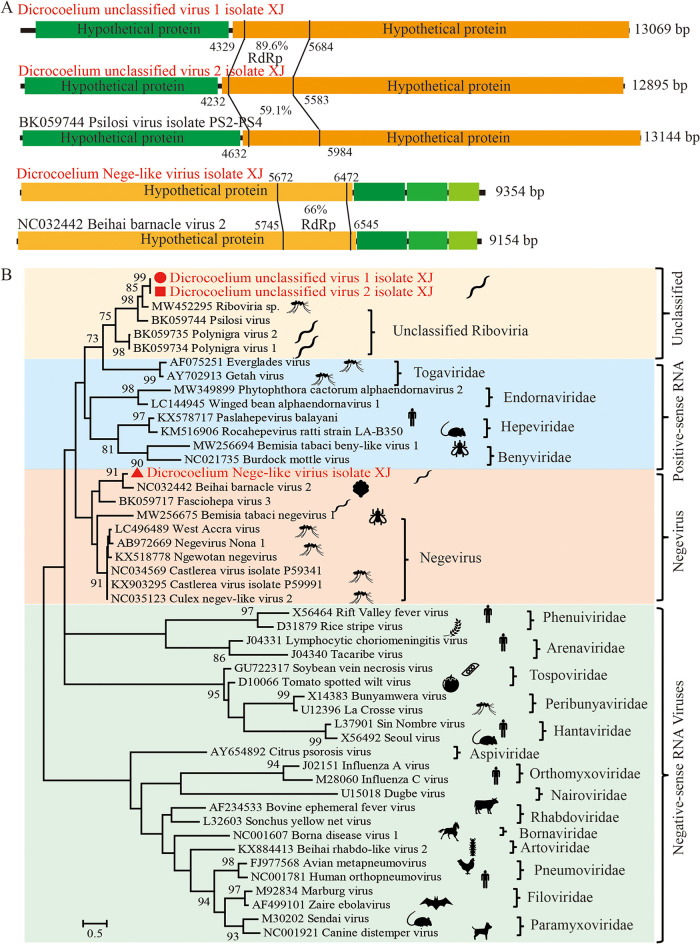
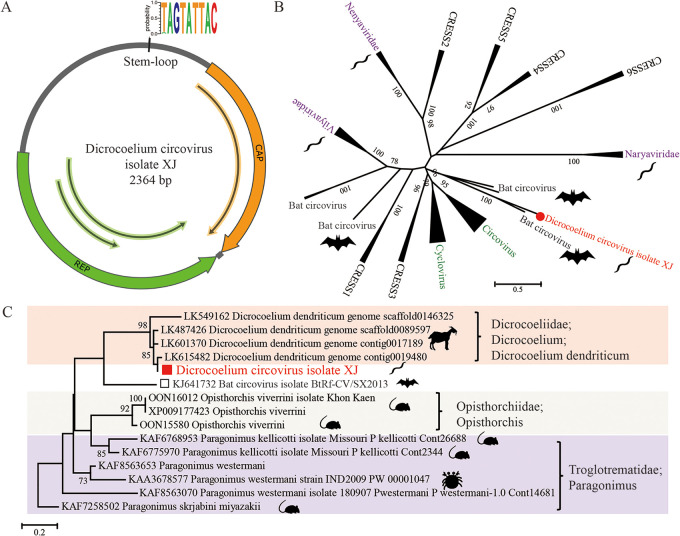
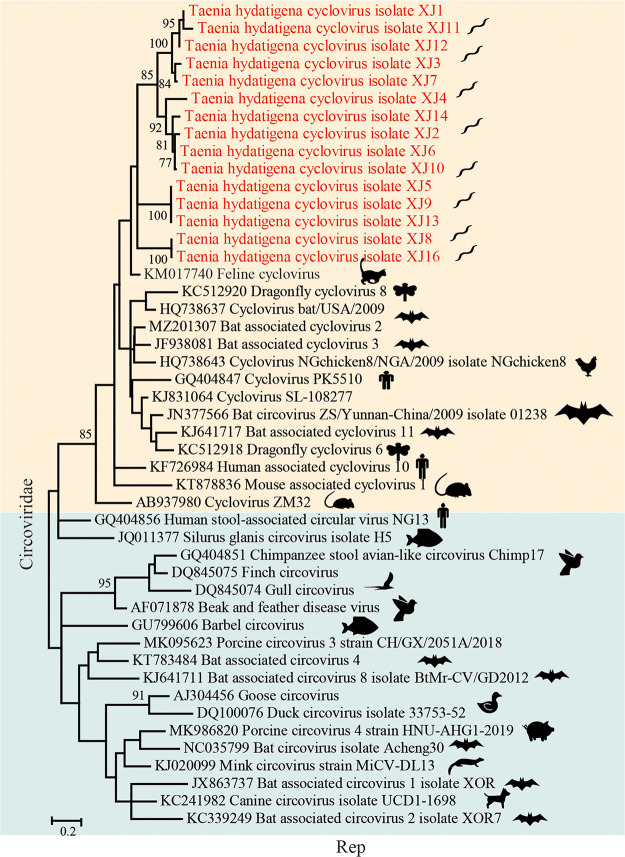
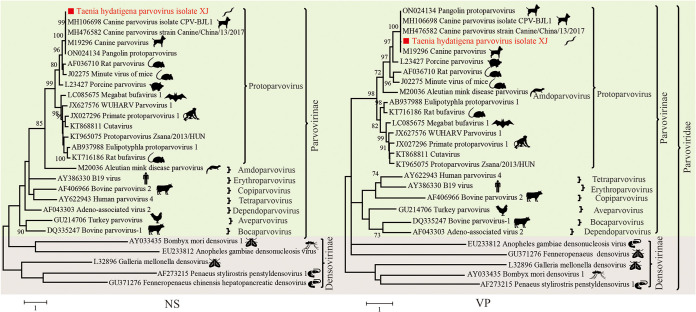
Parasitic flatworms infect diverse vertebrates and are major threats to animal and even human health; however, little is known about the virome of these lower life forms. Using viral metagenomic sequencing, we characterized the virome of the parasitic flatworms collected from major domestic animals, including Dicrocoelium lanceatum and Taenia hydatigena, Echinococcus granulosus sensu stricto and Echinococcus multilocularis. Seven and three different viruses were discovered from D. lanceatum and T. hydatigena, respectively, and no viral sequences were found in adult tapeworms and protoscoleces of E. granulosus sensu stricto and E. multilocularis. Two out of the five parasitic flatworm species carry viruses, showing a host specificity of these viruses. These viruses belong to the Parvoviridae, Circoviridae, unclassified circular, Rep-encoding single-stranded (CRESS) DNA virus, Rhabdoviridae, Endornaviridae, and unclassified RNA viruses. The presence of multiple highly divergent RNA viruses, especially those that cluster with viruses found in marine animals, implies a deep evolutionary history of parasite-associated viruses. In addition, we found viruses with high identity to common pathogens in dogs, including canine circovirus and canine parvovirus 2. The presence of these viruses in the parasites implies that they may infect parasitic flatworms but does not completely exclude the possibility of contamination from host intestinal contents. Furthermore, we demonstrated that certain viruses, such as CRESS DNA virus may integrate into the genome of their host. Our results expand the knowledge of viral diversity in parasites of important domestic animals, highlighting the need for further investigations of their prevalence among other parasites of key animals. IMPORTANCE Characterizing the virome of parasites is important for unveiling the viral diversity, evolution, and ecology and will help to understand the "Russian doll" pattern among viruses, parasites, and host animals. Our data indicate that diverse viruses are present in specific parasitic flatworms, including viruses that may have an ancient evolutionary history and viruses currently circulating in parasite-infected host animals. These data also raise the question of whether parasitic flatworms acquire and/or carry some viruses that may have transmission potential to animals. In addition, through the study of virus-parasite-host interactions, including the influence of viral infection on the life cycle of the parasite, as well as its fitness and pathogenicity to the host, we could find new strategies to prevent and control parasitic diseases.
Keywords: metagenomics; parasitic flatworms; viral metagenomics; virome; virus discovery; virus evolution.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
A World of Viruses Nested within Parasites: Unraveling Viral Diversity within Parasitic Flatworms (Platyhelminthes).Microbiol Spectr. 2022 Jun 29;10(3):e0013822. doi: 10.1128/spectrum.00138-22. Epub 2022 May 10. Microbiol Spectr. 2022. PMID: 35536058 Free PMC article.
-
Virome of crab-eating (Cerdocyon thous) and pampas foxes (Lycalopex gymnocercus) from southern Brazil and Uruguay.Infect Genet Evol. 2020 Nov;85:104421. doi: 10.1016/j.meegid.2020.104421. Epub 2020 Jun 21. Infect Genet Evol. 2020. PMID: 32580027 Free PMC article.
-
Virome of Bat Guano from Nine Northern California Roosts.J Virol. 2021 Jan 13;95(3):e01713-20. doi: 10.1128/JVI.01713-20. Print 2021 Jan 13. J Virol. 2021. PMID: 33115864 Free PMC article.
-
Diversity and evolution of the animal virome.Nat Rev Microbiol. 2022 Jun;20(6):321-334. doi: 10.1038/s41579-021-00665-x. Epub 2022 Jan 4. Nat Rev Microbiol. 2022. PMID: 34983966 Review.
-
Making sense of the virome in light of evolution and ecology.Proc Biol Sci. 2025 Apr;292(2044):20250389. doi: 10.1098/rspb.2025.0389. Epub 2025 Apr 2. Proc Biol Sci. 2025. PMID: 40169018 Free PMC article. Review.
Cited by
-
Diverse RNA viruses of parasitic nematodes can elicit antibody responses in vertebrate hosts.Nat Microbiol. 2024 Oct;9(10):2488-2505. doi: 10.1038/s41564-024-01796-6. Epub 2024 Sep 4. Nat Microbiol. 2024. PMID: 39232205 Free PMC article.
-
A novel picorna-like virus in the flatworm Stenostomum leucops (Catenulida).Arch Virol. 2024 Nov 15;169(12):244. doi: 10.1007/s00705-024-06175-4. Arch Virol. 2024. PMID: 39542977
-
Three Distinct Circovirids Identified in a Tapeworm Recovered from a Bobcat (Lynx rufus).Viruses. 2025 May 23;17(6):745. doi: 10.3390/v17060745. Viruses. 2025. PMID: 40573336 Free PMC article.
-
Rodent Gut Bacteria Coexisting with an Insect Gut Virus in Parasitic Cysts: Metagenomic Evidence of Microbial Translocation and Co-adaptation in Spatially-Confined Niches.bioRxiv [Preprint]. 2024 Mar 23:2024.03.22.585885. doi: 10.1101/2024.03.22.585885. bioRxiv. 2024. Update in: Microorganisms. 2024 May 31;12(6):1130. doi: 10.3390/microorganisms12061130. PMID: 38562820 Free PMC article. Updated. Preprint.
-
Rodent Gut Bacteria Coexisting with an Insect Gut Virus in Tapeworm Parasitic Cysts: Metagenomic Evidence of Microbial Selection in Extra-Intestinal Clinical Niches.Microorganisms. 2024 May 31;12(6):1130. doi: 10.3390/microorganisms12061130. Microorganisms. 2024. PMID: 38930512 Free PMC article.
References
-
- Erkyihun GA, Alemayehu MB. 2022. One Health approach for the control of zoonotic diseases. Zoonoses 2. doi: 10.15212/ZOONOSES-2022-0037. - DOI
-
- Guha Dharmarajan RL, Chanda E, Dean KR, Dirzo R, Jakobsen KS, Khan I, Leirs H, Shi Z-L, Wolfe ND, Yang R, Stenseth NC. 2022. The animal origin of major human infectious diseases: what can past epidemics teach us about preventing the next pandemic? Zoonoses 2. doi: 10.15212/ZOONOSES-2021-0028. - DOI
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
