

Identification of hidden associations among eukaryotic genes through statistical analysis of coevolutionary transitions
- PMID: 37043529
- PMCID: PMC10120013
- DOI: 10.1073/pnas.2218329120
Identification of hidden associations among eukaryotic genes through statistical analysis of coevolutionary transitions
Abstract
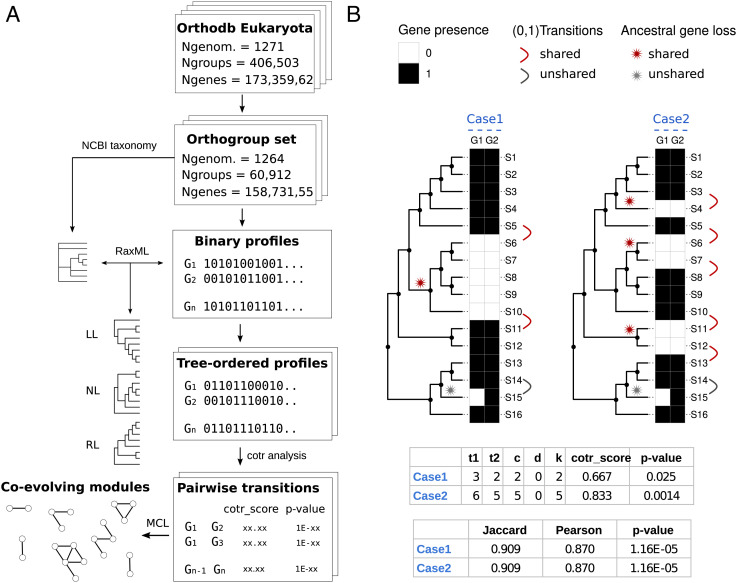
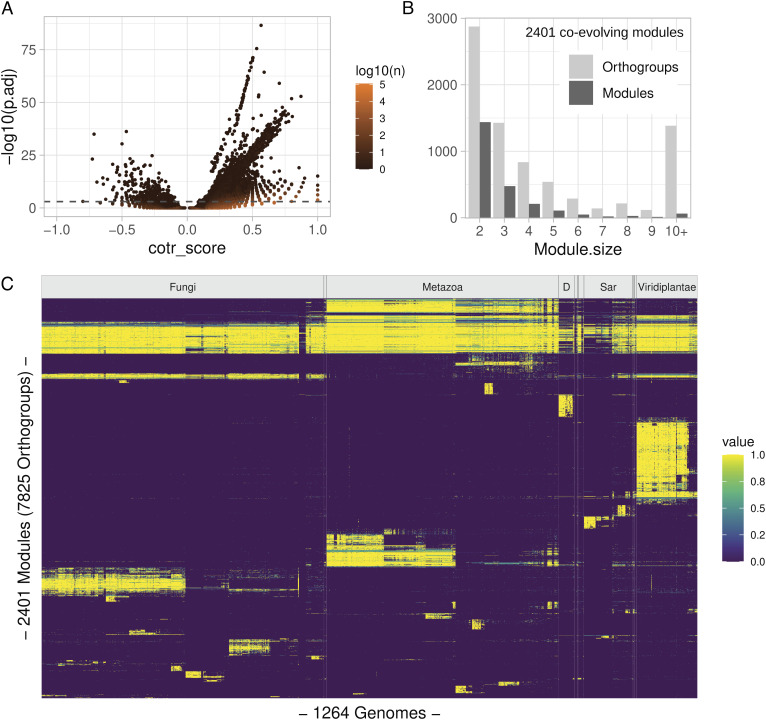
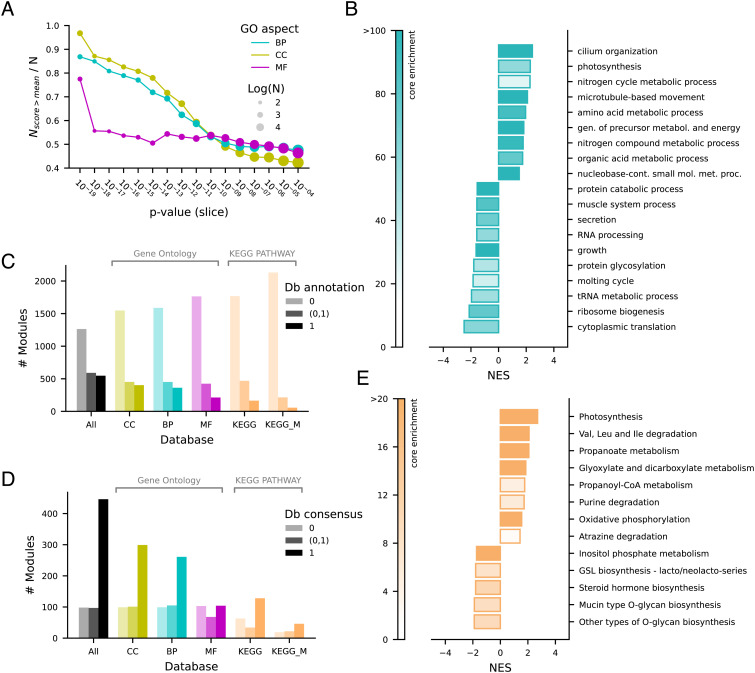
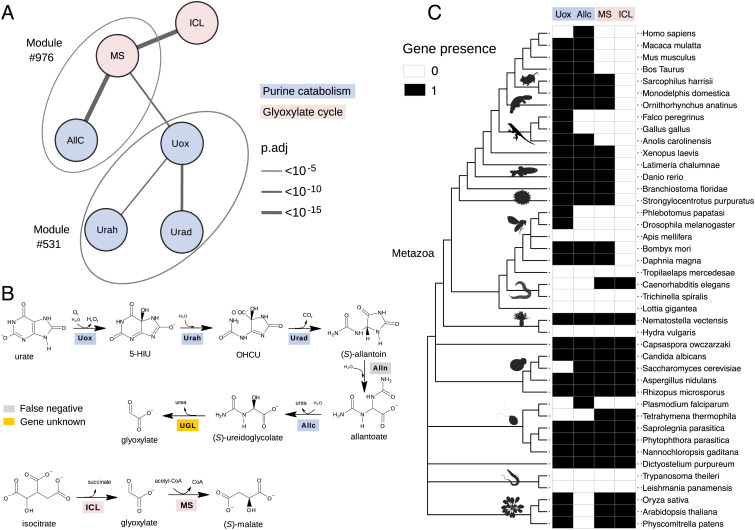
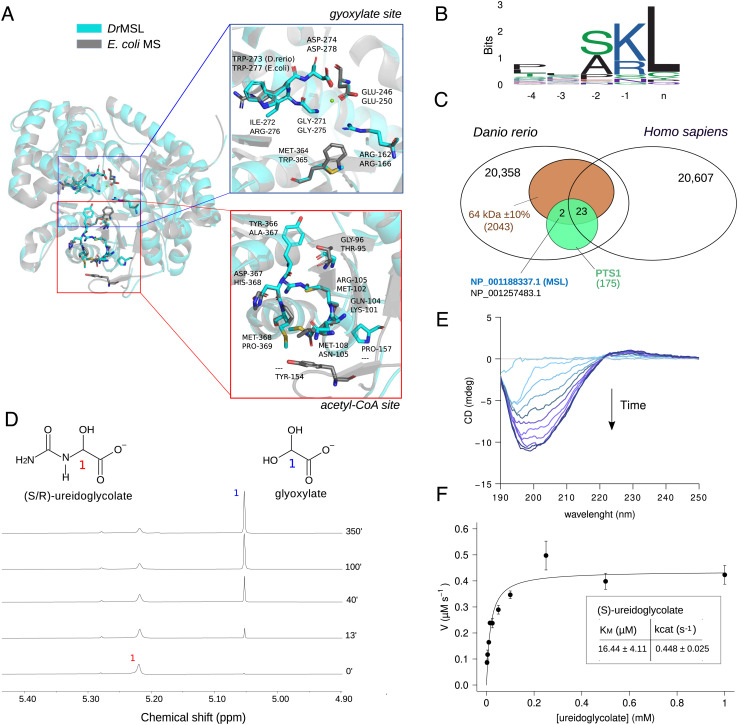
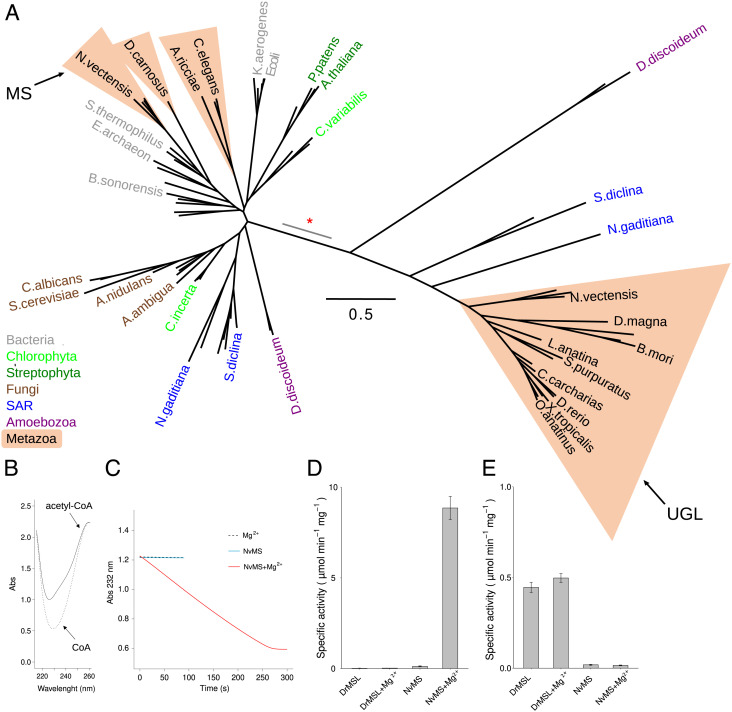
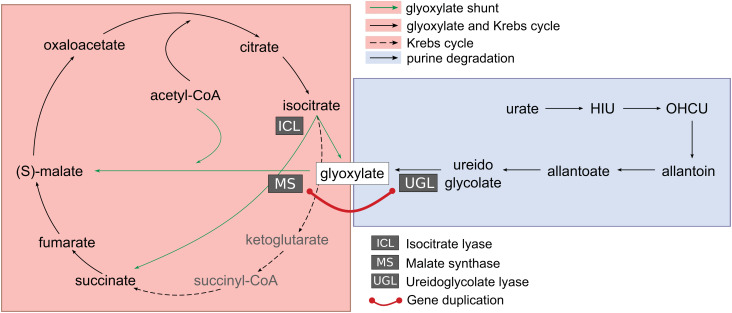
Coevolution at the gene level, as reflected by correlated events of gene loss or gain, can be revealed by phylogenetic profile analysis. The optimal method and metric for comparing phylogenetic profiles, especially in eukaryotic genomes, are not yet established. Here, we describe a procedure suitable for large-scale analysis, which can reveal coevolution based on the assessment of the statistical significance of correlated presence/absence transitions between gene pairs. This metric can identify coevolution in profiles with low overall similarities and is not affected by similarities lacking coevolutionary information. We applied the procedure to a large collection of 60,912 orthologous gene groups (orthogroups) in 1,264 eukaryotic genomes extracted from OrthoDB. We found significant cotransition scores for 7,825 orthogroups associated in 2,401 coevolving modules linking known and unknown genes in protein complexes and biological pathways. To demonstrate the ability of the method to predict hidden gene associations, we validated through experiments the involvement of vertebrate malate synthase-like genes in the conversion of (S)-ureidoglycolate into glyoxylate and urea, the last step of purine catabolism. This identification explains the presence of glyoxylate cycle genes in metazoa and suggests an anaplerotic role of purine degradation in early eukaryotes.
Keywords: coevolution; gene association; glyoxylate cycle; purine catabolism; statistical significance.
Conflict of interest statement
The authors declare no competing interest.
Figures
Comment in
-
Phylogenetic profiling in eukaryotes comes of age.Proc Natl Acad Sci U S A. 2023 May 9;120(19):e2305013120. doi: 10.1073/pnas.2305013120. Epub 2023 May 1. Proc Natl Acad Sci U S A. 2023. PMID: 37126713 Free PMC article. No abstract available.
Similar articles
-
Phylogenetic profiling in eukaryotes: The effect of species, orthologous group, and interactome selection on protein interaction prediction.PLoS One. 2022 Apr 14;17(4):e0251833. doi: 10.1371/journal.pone.0251833. eCollection 2022. PLoS One. 2022. PMID: 35421089 Free PMC article.
-
Giant Viruses Encode Actin-Related Proteins.Mol Biol Evol. 2022 Feb 3;39(2):msac022. doi: 10.1093/molbev/msac022. Mol Biol Evol. 2022. PMID: 35150280 Free PMC article.
-
Analysis of coevolving gene families using mutually exclusive orthologous modules.Genome Biol Evol. 2011;3:413-23. doi: 10.1093/gbe/evr030. Epub 2011 Apr 17. Genome Biol Evol. 2011. PMID: 21498882 Free PMC article.
-
Origin and evolution of spliceosomal introns.Biol Direct. 2012 Apr 16;7:11. doi: 10.1186/1745-6150-7-11. Biol Direct. 2012. PMID: 22507701 Free PMC article. Review.
-
Patterns and impacts of nonvertical evolution in eukaryotes: a paradigm shift.Ann N Y Acad Sci. 2020 Sep;1476(1):78-92. doi: 10.1111/nyas.14471. Epub 2020 Aug 28. Ann N Y Acad Sci. 2020. PMID: 32860228 Free PMC article. Review.
Cited by
-
Quest for Orthologs in the Era of Biodiversity Genomics.Genome Biol Evol. 2024 Oct 9;16(10):evae224. doi: 10.1093/gbe/evae224. Genome Biol Evol. 2024. PMID: 39404012 Free PMC article. Review.
-
Cysteine Enrichment Mediates Co-Option of Uricase in Reptilian Skin and Transition to Uricotelism.Mol Biol Evol. 2023 Sep 1;40(9):msad200. doi: 10.1093/molbev/msad200. Mol Biol Evol. 2023. PMID: 37695804 Free PMC article.
-
A CUG-initiated CATSPERθ functions in the CatSper channel assembly and serves as a checkpoint for flagellar trafficking.Proc Natl Acad Sci U S A. 2023 Sep 26;120(39):e2304409120. doi: 10.1073/pnas.2304409120. Epub 2023 Sep 19. Proc Natl Acad Sci U S A. 2023. PMID: 37725640 Free PMC article.
-
C11orf54 catalyzes L-xylulose formation in human metabolism.Proc Natl Acad Sci U S A. 2025 Aug 5;122(31):e2506597122. doi: 10.1073/pnas.2506597122. Epub 2025 Jul 30. Proc Natl Acad Sci U S A. 2025. PMID: 40737316
-
Phylogenetic profiling in eukaryotes comes of age.Proc Natl Acad Sci U S A. 2023 May 9;120(19):e2305013120. doi: 10.1073/pnas.2305013120. Epub 2023 May 1. Proc Natl Acad Sci U S A. 2023. PMID: 37126713 Free PMC article. No abstract available.
References
-
- de Juan D., Pazos F., Valencia A., Emerging methods in protein co-evolution. Nat. Rev. Genet. 14, 249–261 (2013). - PubMed
-
- Ebert D., Fields P. D., Host–parasite co-evolution and its genomic signature. Nat. Rev. Genet. 21, 754–768 (2020). - PubMed
-
- Tatusov R. L., Koonin E. V., Lipman D. J., A genomic perspective on protein families. Science 278, 631–637 (1997). - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
