

Antisense oligonucleotide therapy for H3.3K27M diffuse midline glioma
- PMID: 37043556
- PMCID: PMC10263181
- DOI: 10.1126/scitranslmed.add8280
Antisense oligonucleotide therapy for H3.3K27M diffuse midline glioma
Abstract
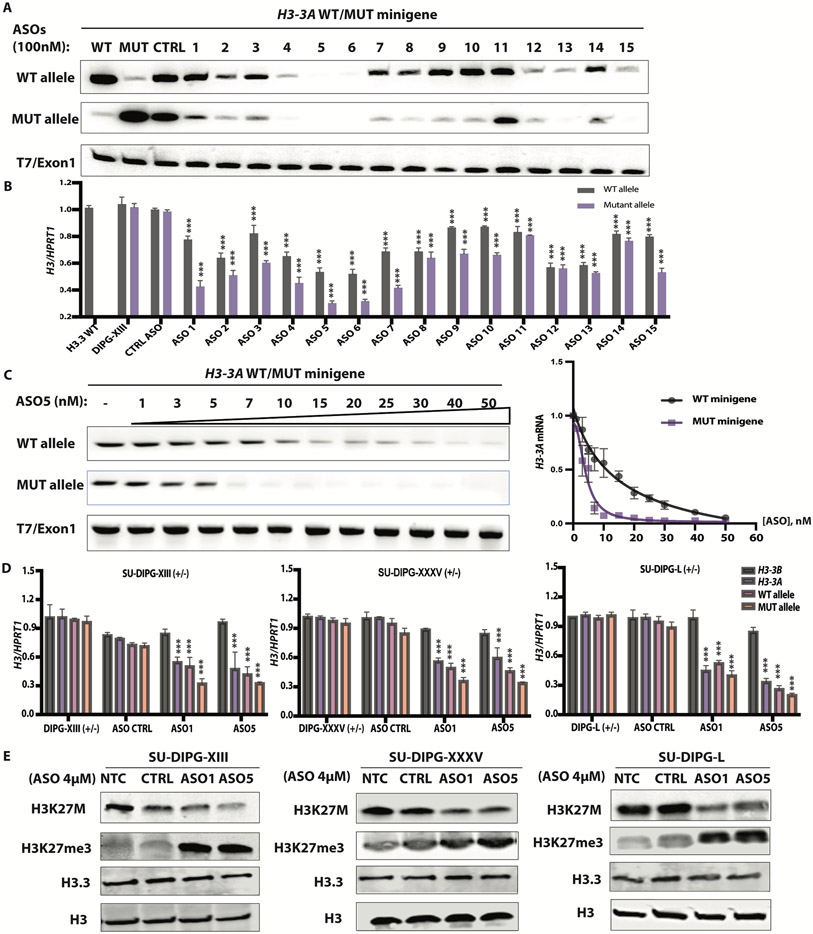
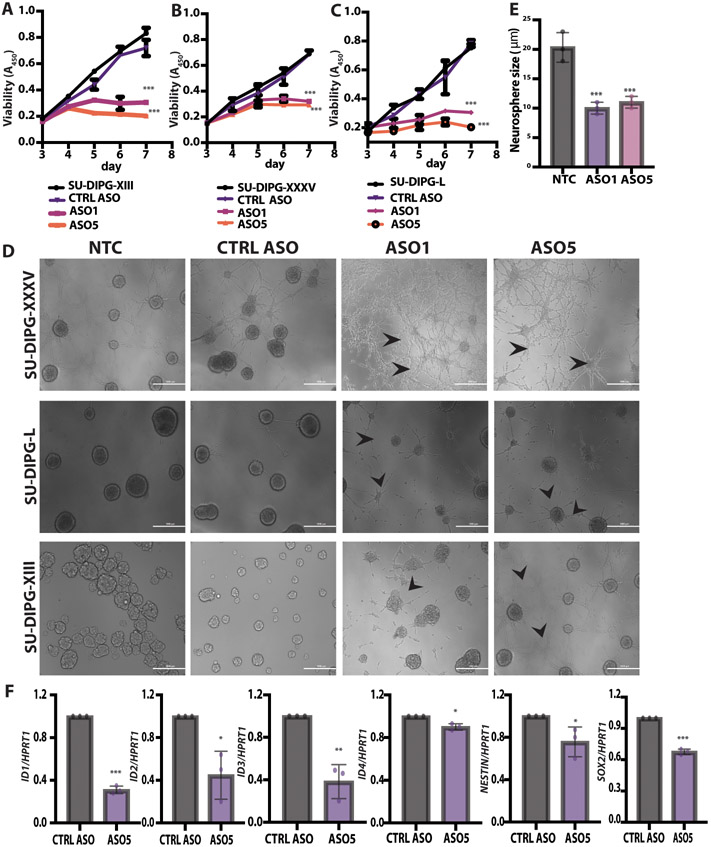
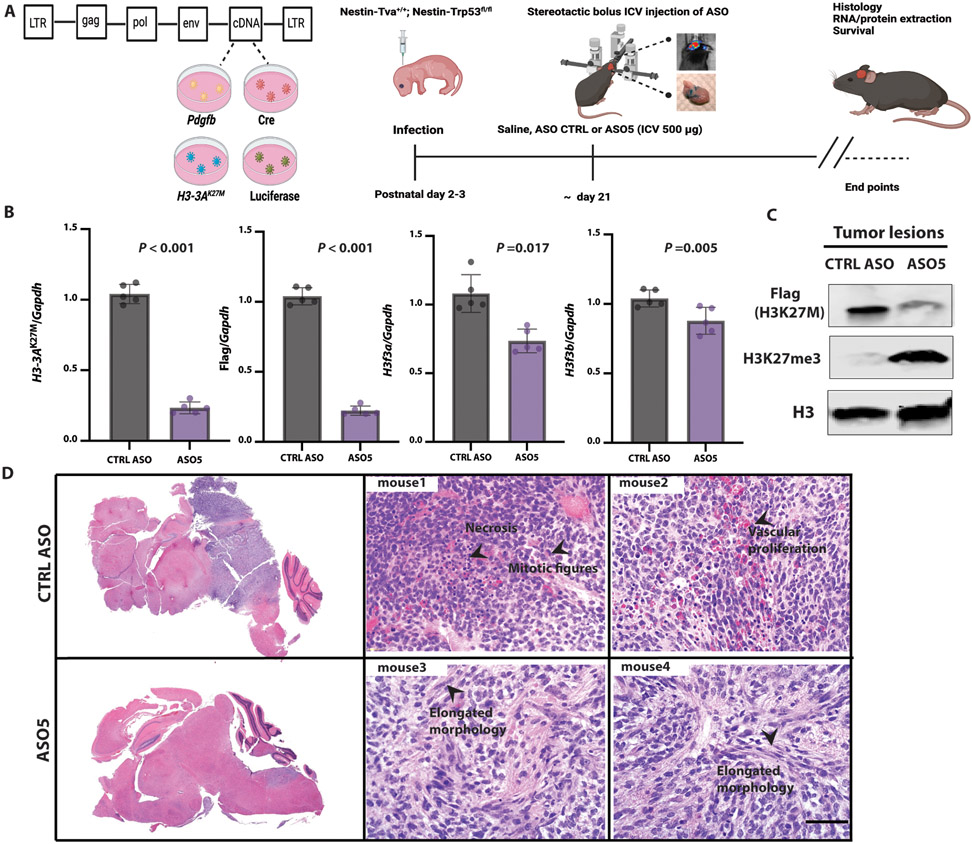
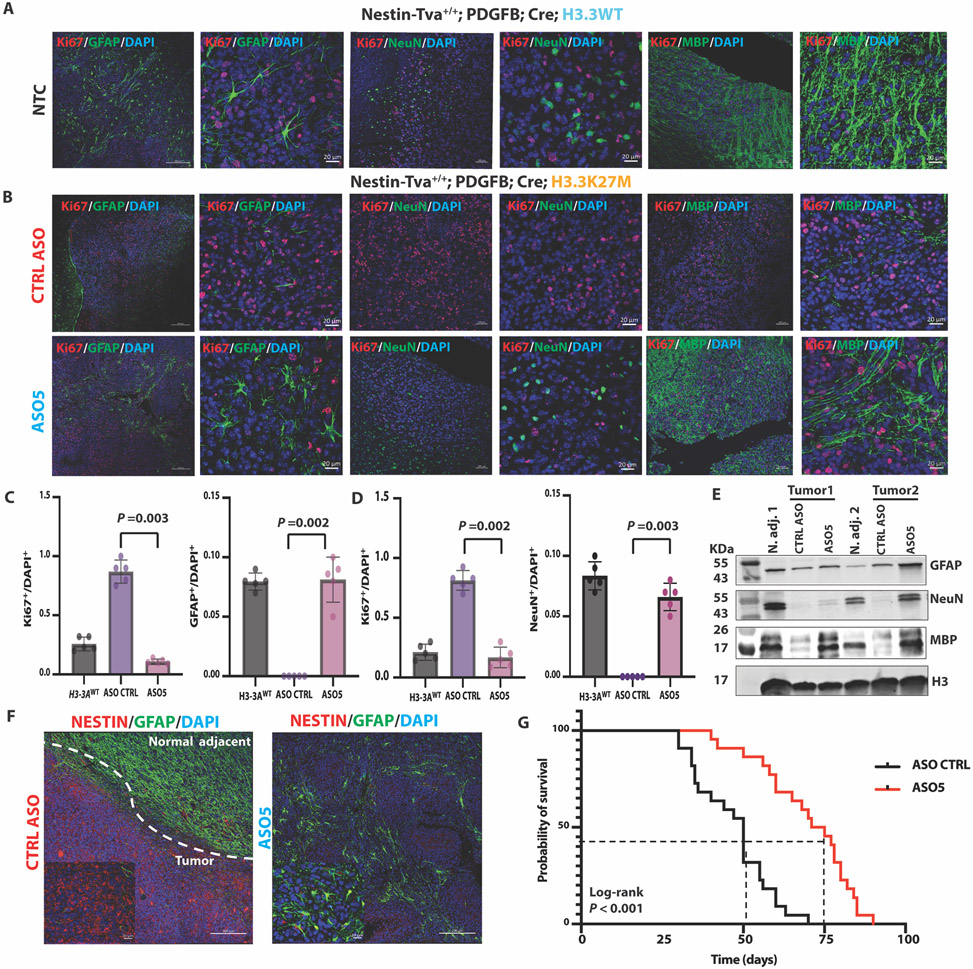
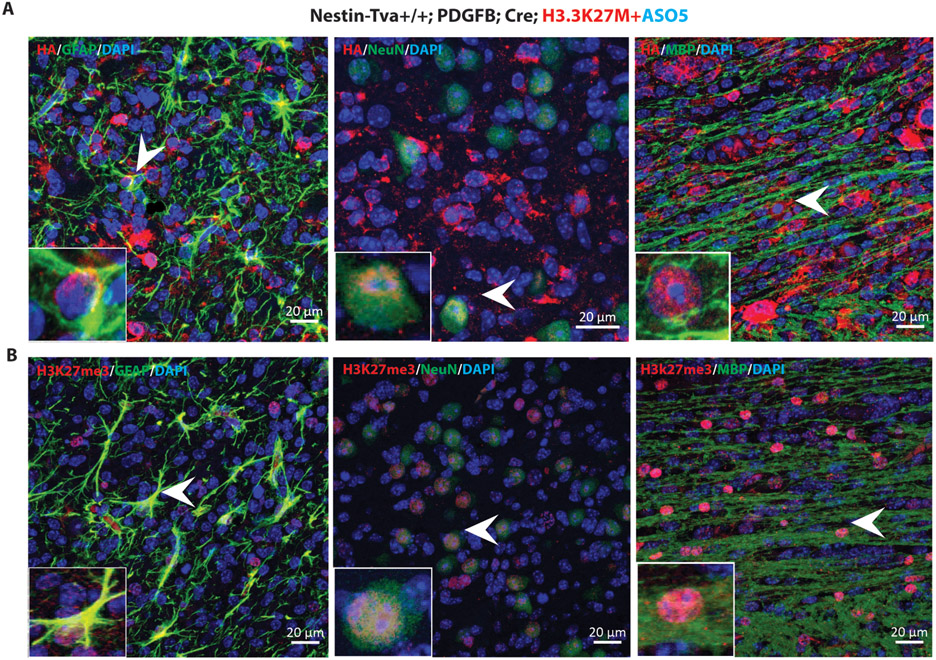
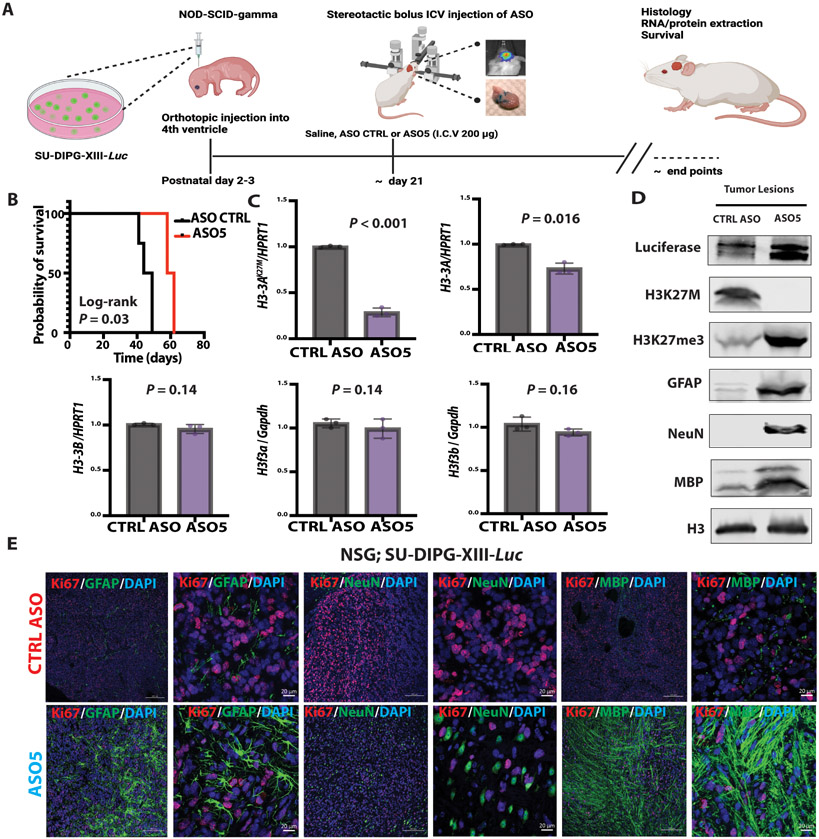
Diffuse midline gliomas (DMGs) are pediatric high-grade brain tumors in the thalamus, midbrain, or pons; the latter subgroup are termed diffuse intrinsic pontine gliomas (DIPG). The brain stem location of these tumors limits the clinical management of DIPG, resulting in poor outcomes for patients. A heterozygous, somatic point mutation in one of two genes coding for the noncanonical histone H3.3 is present in most DIPG tumors. This dominant mutation in the H3-3A gene results in replacement of lysine 27 with methionine (K27M) and causes a global reduction of trimethylation on K27 of all wild-type histone H3 proteins, which is thought to be a driving event in gliomagenesis. In this study, we designed and systematically screened 2'-O-methoxyethyl phosphorothioate antisense oligonucleotides (ASOs) that direct RNase H-mediated knockdown of H3-3A mRNA. We identified a lead ASO that effectively reduced H3-3A mRNA and H3.3K27M protein and restored global H3K27 trimethylation in patient-derived neurospheres. We then tested the lead ASO in two mouse models of DIPG: an immunocompetent mouse model using transduced mutant human H3-3A cDNA and an orthotopic xenograft with patient-derived cells. In both models, ASO treatment restored K27 trimethylation of histone H3 proteins and reduced tumor growth, promoted neural stem cell differentiation into astrocytes, neurons, and oligodendrocytes, and increased survival. These results demonstrate the involvement of the H3.3K27M oncohistone in tumor maintenance, confirm the reversibility of the aberrant epigenetic changes it promotes, and provide preclinical proof of concept for DMG antisense therapy.
Figures
Comment in
-
Antisense oligonucleotide slows glioma growth.Nat Rev Drug Discov. 2023 Jun;22(6):445. doi: 10.1038/d41573-023-00077-x. Nat Rev Drug Discov. 2023. PMID: 37165091 No abstract available.
-
Promising results for antisense RNA therapy in mouse models of diffuse midline glioma.Nat Rev Neurol. 2023 Jul;19(7):386. doi: 10.1038/s41582-023-00829-8. Nat Rev Neurol. 2023. PMID: 37253852 No abstract available.
References
-
- Harutyunyan AS, Krug B, Chen H, Papillon-Cavanagh S, Zeinieh M, De Jay N, Deshmukh S, Chen CCL, Belle J, Mikael LG, Marchione DM, Li R, Nikbakht H, Hu B, Cagnone G, Cheung WA, Mohammadnia A, Bechet D, Faury D, McConechy MK, Pathania M, Jain SU, Ellezam B, Weil AG, Montpetit A, Salomoni P, Pastinen T, Lu C, Lewis PW, Garcia BA, Kleinman CL, Jabado N, Majewski J, H3K27M induces defective chromatin spread of PRC2-mediated repressive H3K27me2/me3 and is essential for glioma tumorigenesis. Nat. Commun 10, 1262 (2019) - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
