

Genomic Diagnosis of Rare Pediatric Disease in the United Kingdom and Ireland
- PMID: 37043637
- PMCID: PMC7614484
- DOI: 10.1056/NEJMoa2209046
Genomic Diagnosis of Rare Pediatric Disease in the United Kingdom and Ireland
Abstract
Background: Pediatric disorders include a range of highly penetrant, genetically heterogeneous conditions amenable to genomewide diagnostic approaches. Finding a molecular diagnosis is challenging but can have profound lifelong benefits.
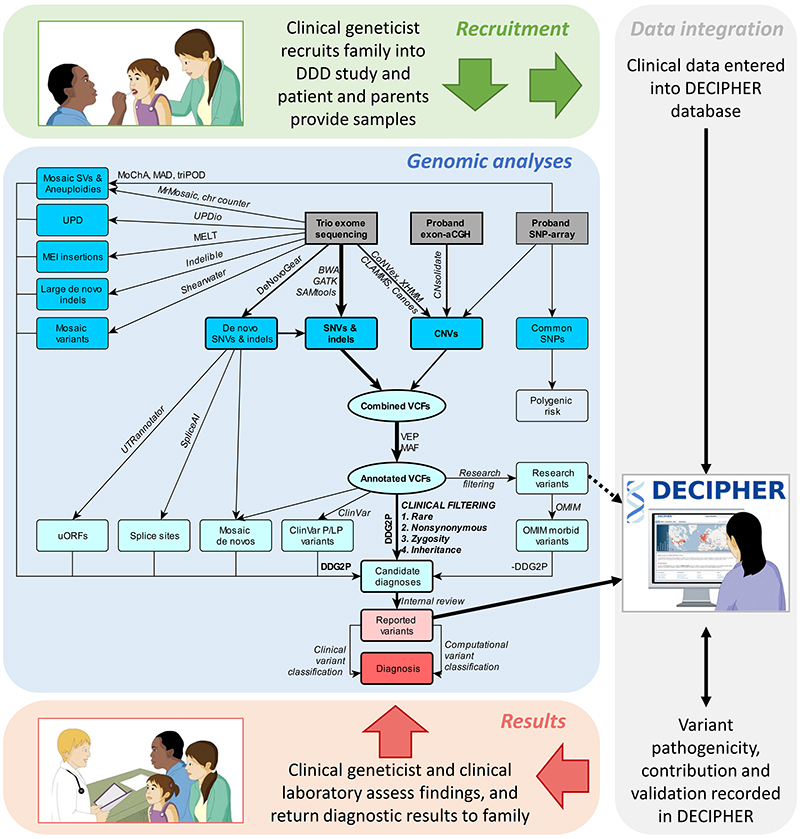
Methods: We conducted a large-scale sequencing study involving more than 13,500 families with probands with severe, probably monogenic, difficult-to-diagnose developmental disorders from 24 regional genetics services in the United Kingdom and Ireland. Standardized phenotypic data were collected, and exome sequencing and microarray analyses were performed to investigate novel genetic causes. We developed an iterative variant analysis pipeline and reported candidate variants to clinical teams for validation and diagnostic interpretation to inform communication with families. Multiple regression analyses were performed to evaluate factors affecting the probability of diagnosis.
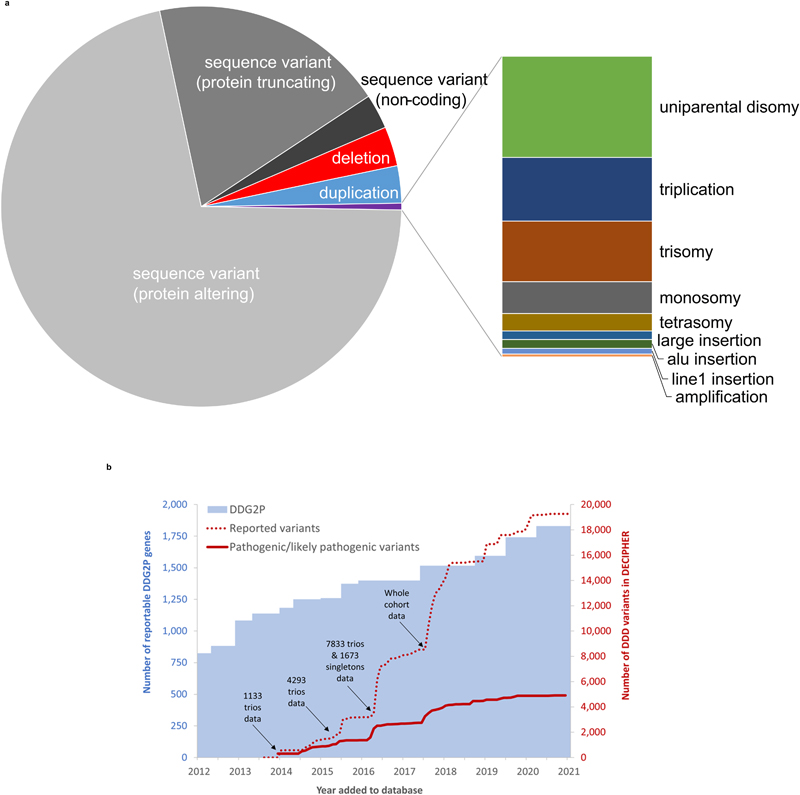
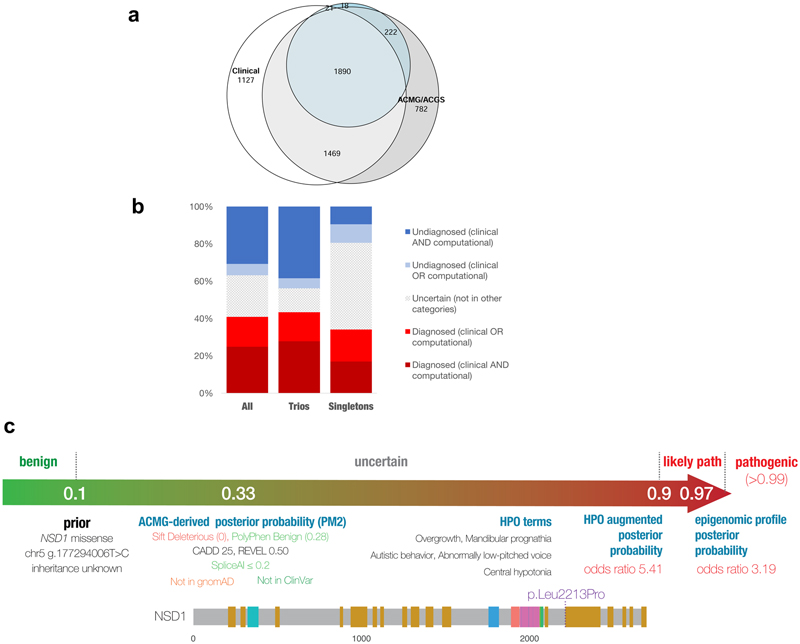
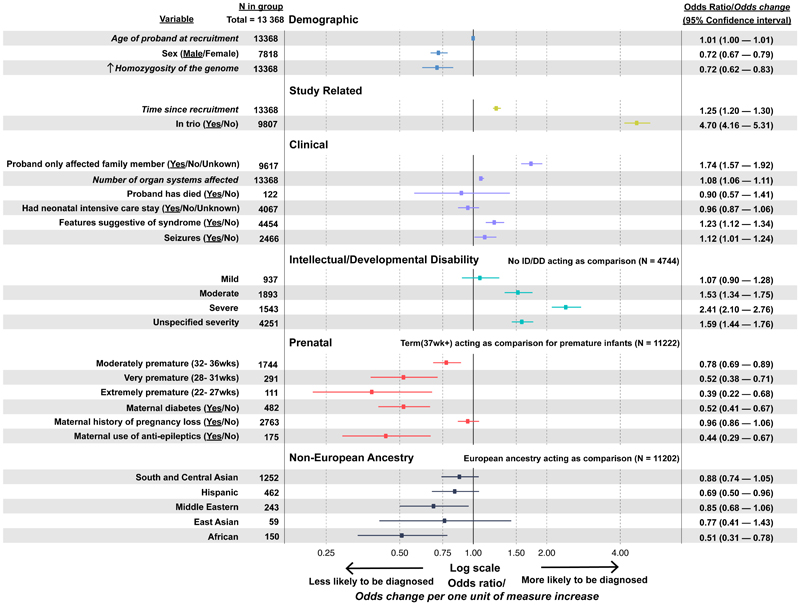
Results: A total of 13,449 probands were included in the analyses. On average, we reported 1.0 candidate variant per parent-offspring trio and 2.5 variants per singleton proband. Using clinical and computational approaches to variant classification, we made a diagnosis in approximately 41% of probands (5502 of 13,449). Of 3599 probands in trios who received a diagnosis by clinical assertion, approximately 76% had a pathogenic de novo variant. Another 22% of probands (2997 of 13,449) had variants of uncertain significance in genes that were strongly linked to monogenic developmental disorders. Recruitment in a parent-offspring trio had the largest effect on the probability of diagnosis (odds ratio, 4.70; 95% confidence interval [CI], 4.16 to 5.31). Probands were less likely to receive a diagnosis if they were born extremely prematurely (i.e., 22 to 27 weeks' gestation; odds ratio, 0.39; 95% CI, 0.22 to 0.68), had in utero exposure to antiepileptic medications (odds ratio, 0.44; 95% CI, 0.29 to 0.67), had mothers with diabetes (odds ratio, 0.52; 95% CI, 0.41 to 0.67), or were of African ancestry (odds ratio, 0.51; 95% CI, 0.31 to 0.78).
Conclusions: Among probands with severe, probably monogenic, difficult-to-diagnose developmental disorders, multimodal analysis of genomewide data had good diagnostic power, even after previous attempts at diagnosis. (Funded by the Health Innovation Challenge Fund and Wellcome Sanger Institute.).
Copyright © 2023 Massachusetts Medical Society.
Figures
References
-
- Adams DR, Eng CM. Next-Generation Sequencing to Diagnose Suspected Genetic Disorders. N Engl J Med. 2018;379(14):1353–62. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical