

The APE2 nuclease is essential for DNA double-strand break repair by microhomology-mediated end joining
- PMID: 37044098
- PMCID: PMC10164096
- DOI: 10.1016/j.molcel.2023.03.017
The APE2 nuclease is essential for DNA double-strand break repair by microhomology-mediated end joining
Abstract
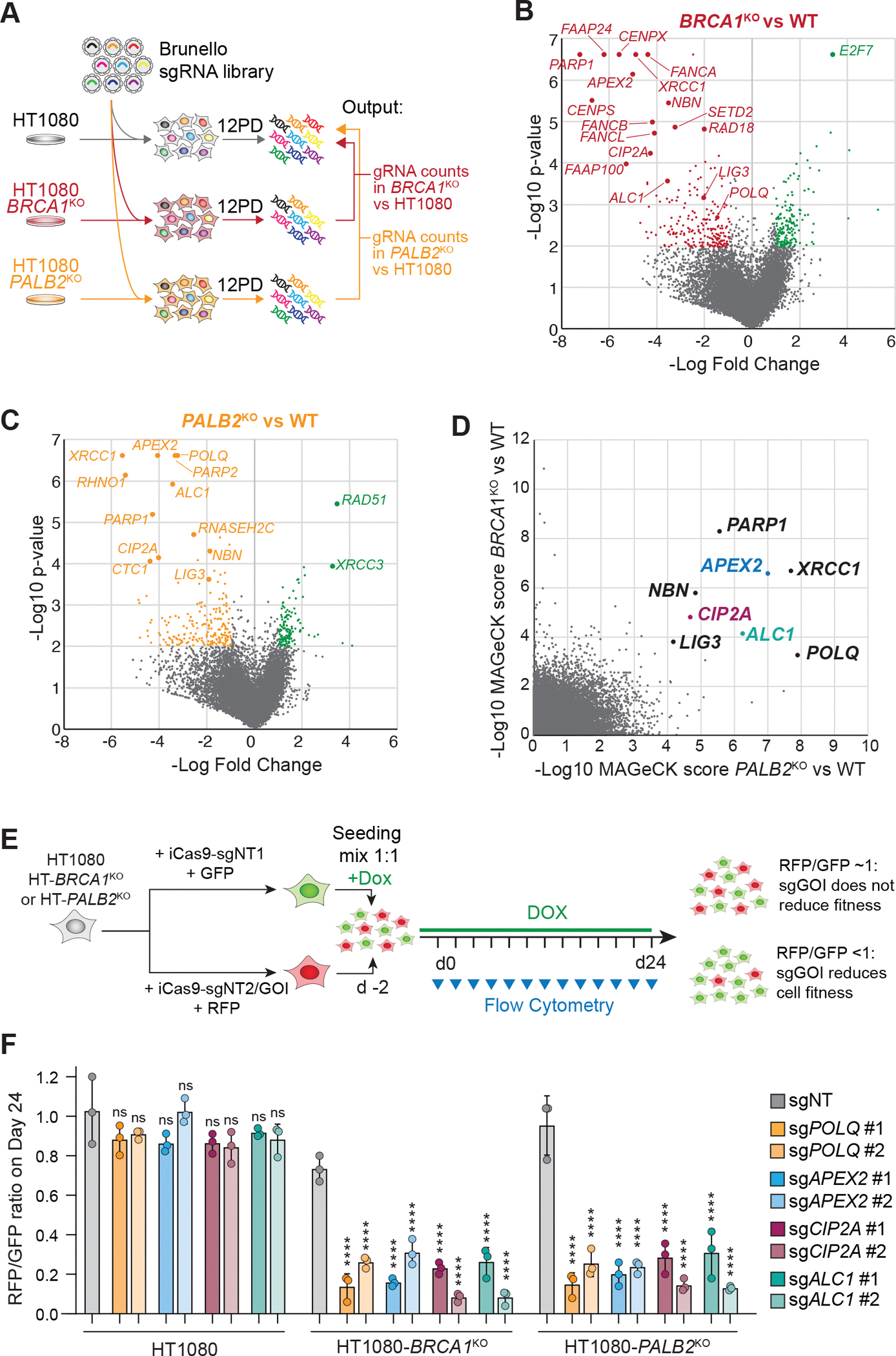
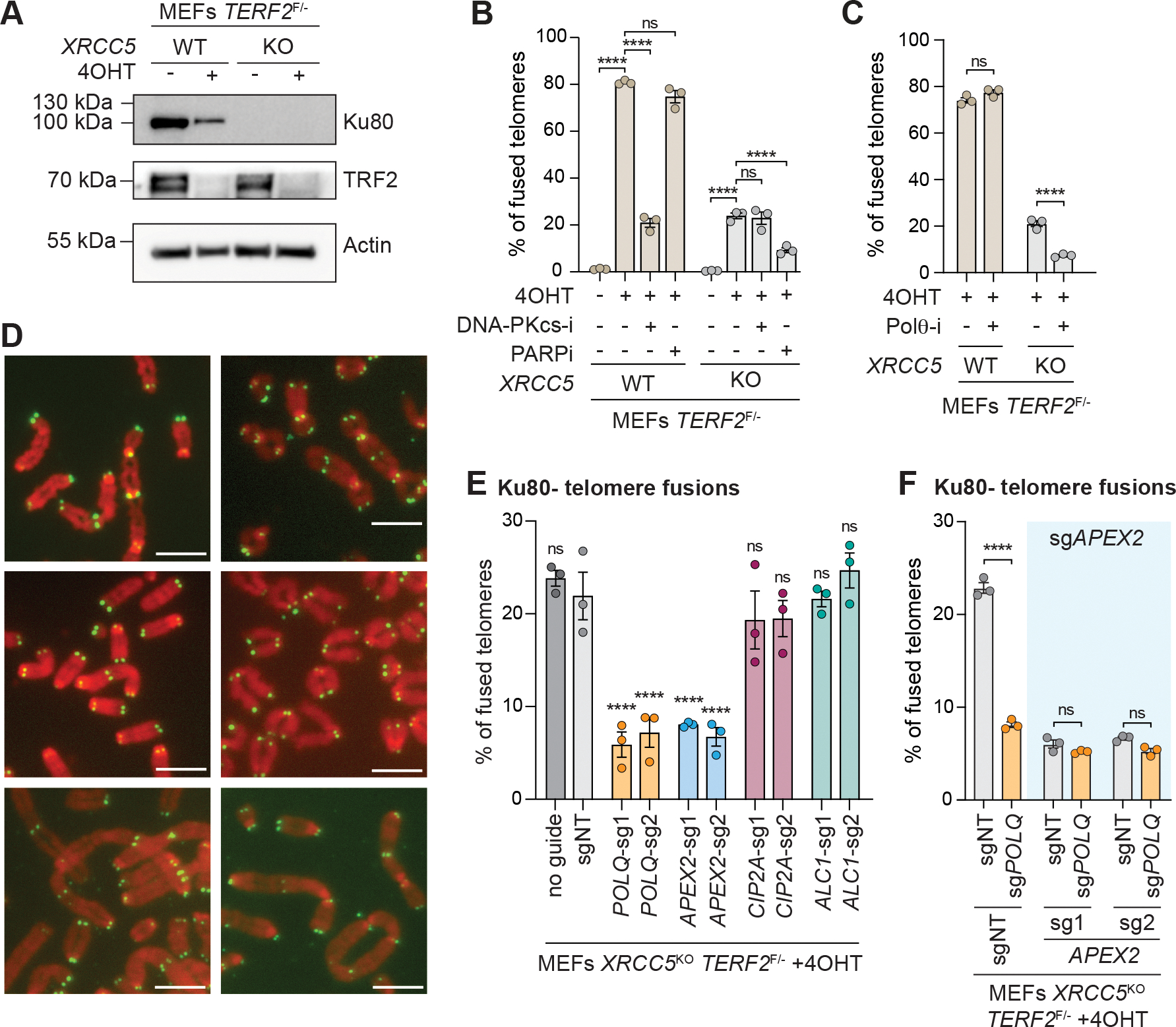
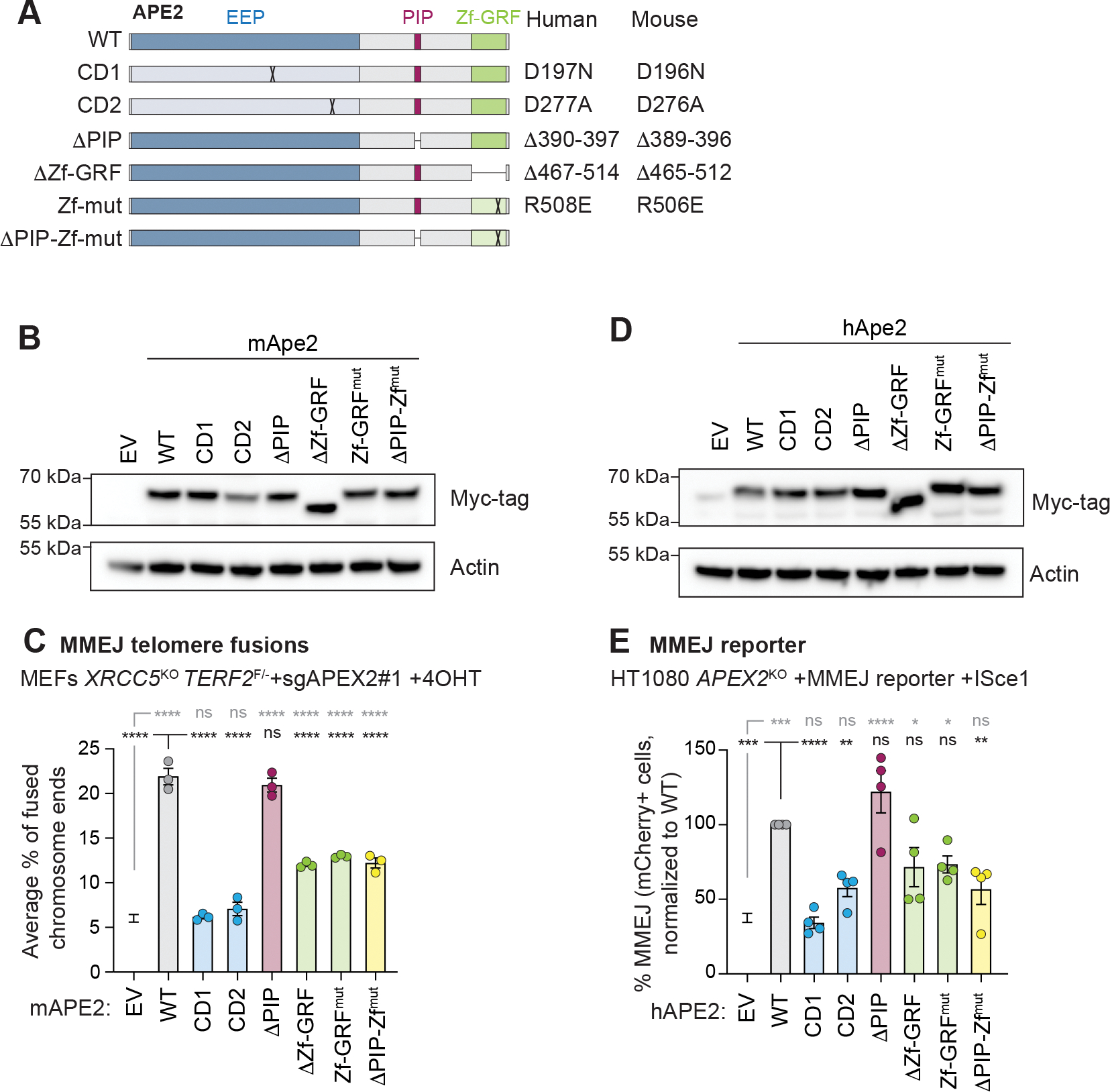
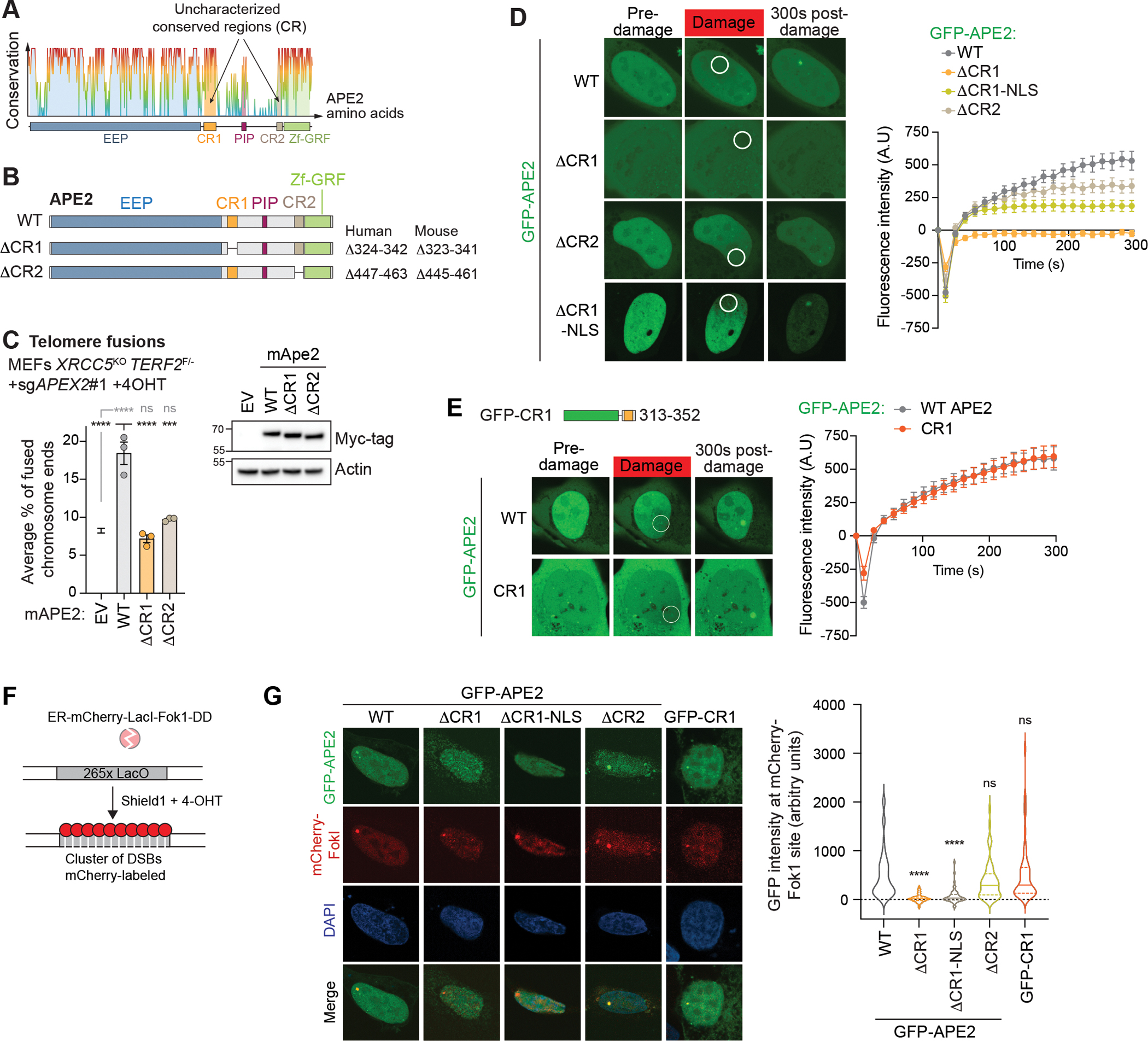
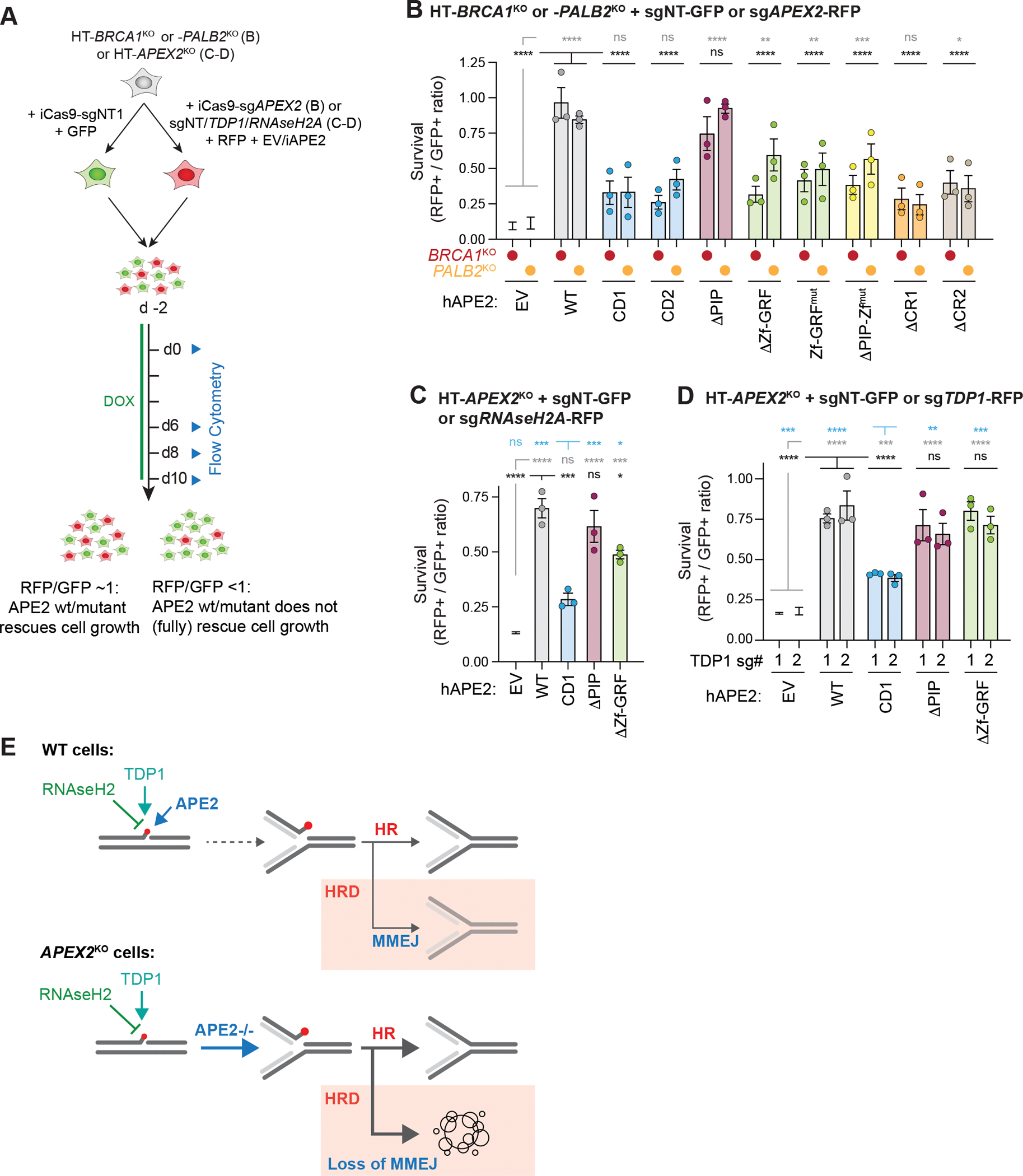
Microhomology-mediated end joining (MMEJ) is an intrinsically mutagenic pathway of DNA double-strand break (DSB) repair essential for proliferation of homologous recombination (HR)-deficient tumors. Although targeting MMEJ has emerged as a powerful strategy to eliminate HR-deficient (HRD) cancers, this is limited by an incomplete understanding of the mechanism and factors required for MMEJ repair. Here, we identify the APE2 nuclease as an MMEJ effector. We show that loss of APE2 inhibits MMEJ at deprotected telomeres and at intra-chromosomal DSBs and is epistatic with Pol Theta for MMEJ activity. Mechanistically, we demonstrate that APE2 possesses intrinsic flap-cleaving activity, that its MMEJ function in cells depends on its nuclease activity, and further identify an uncharacterized domain required for its recruitment to DSBs. We conclude that this previously unappreciated role of APE2 in MMEJ contributes to the addiction of HRD cells to APE2, which could be exploited in the treatment of cancer.
Keywords: APE2; APEX2; DNA repair; HR-deficient; Homologous Recombination; MMEJ; PARP; cancer; synthetic lethality.
Copyright © 2023 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests S.J.B. is co-founder and VP Science Strategy at Artios Pharma Ltd.
Figures


References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
