

Characterization of genome-wide STR variation in 6487 human genomes
- PMID: 37045857
- PMCID: PMC10097659
- DOI: 10.1038/s41467-023-37690-8
Characterization of genome-wide STR variation in 6487 human genomes
Abstract
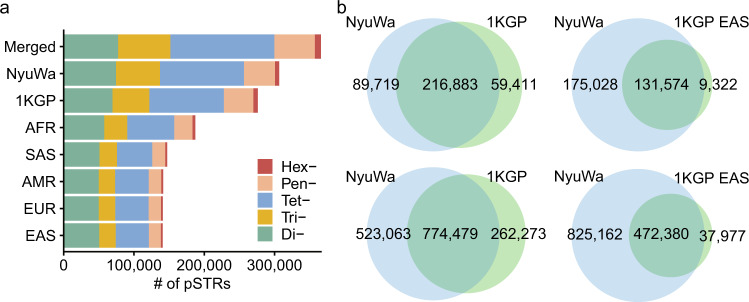
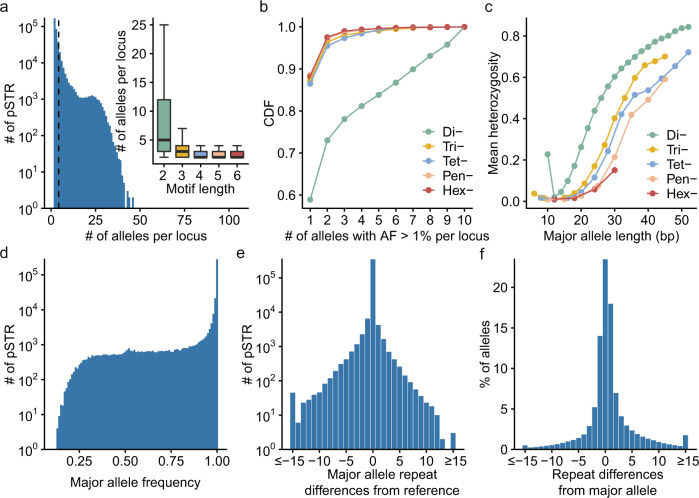
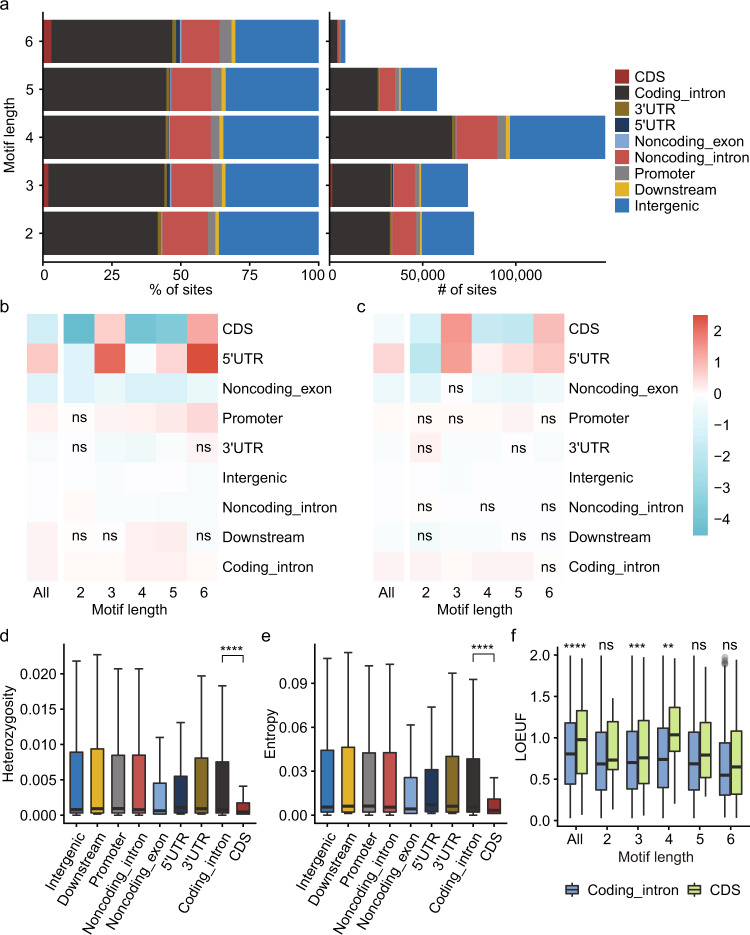
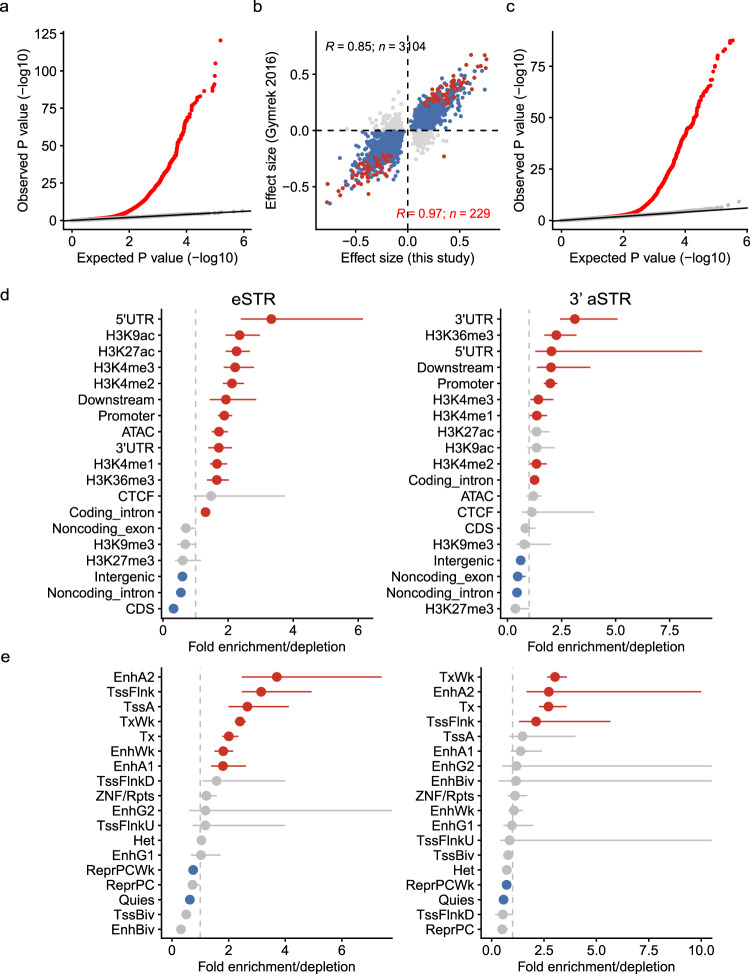
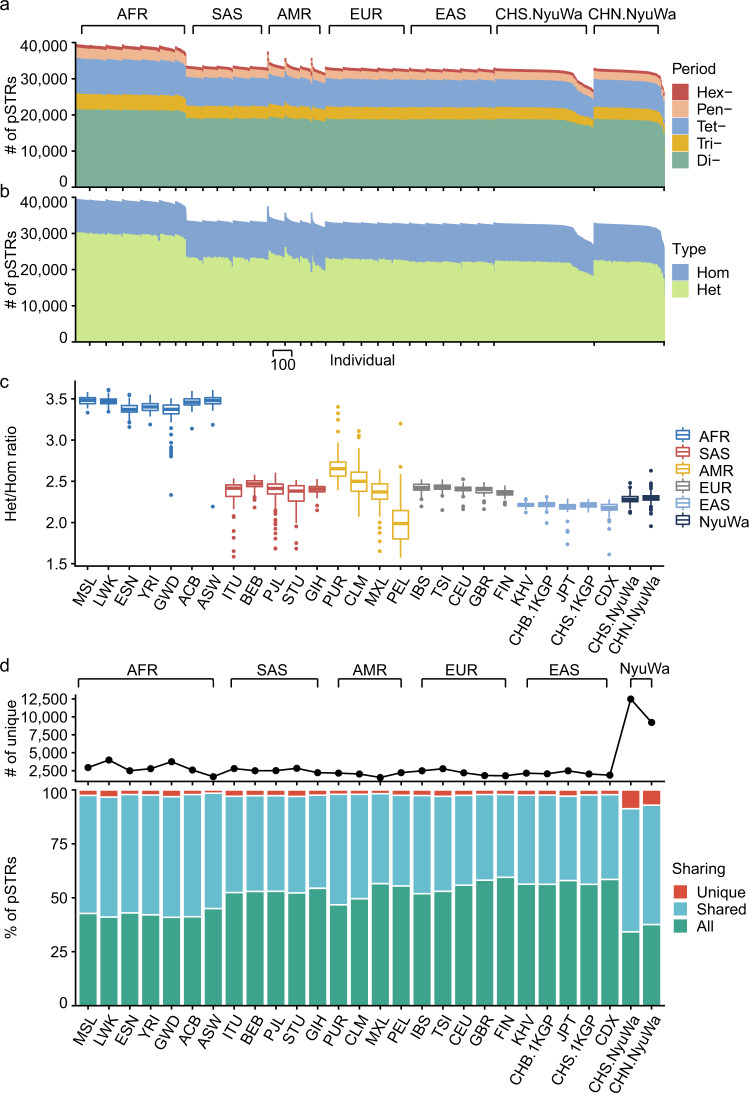
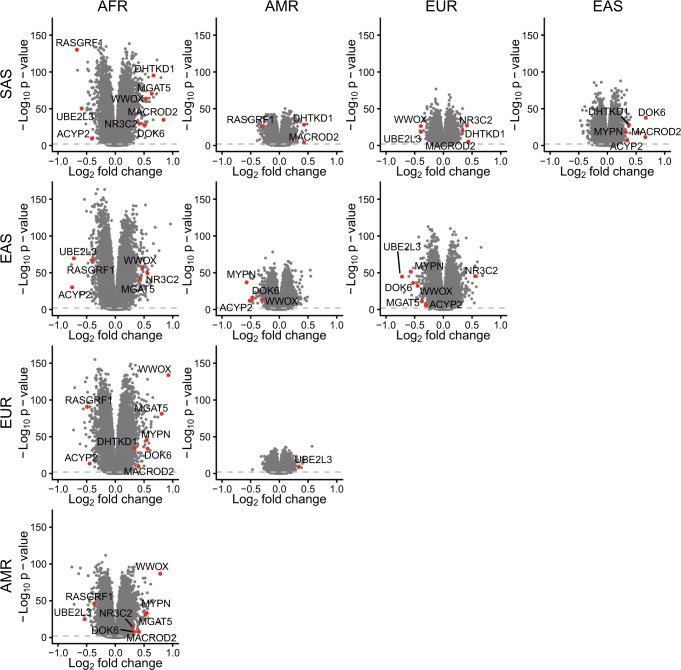
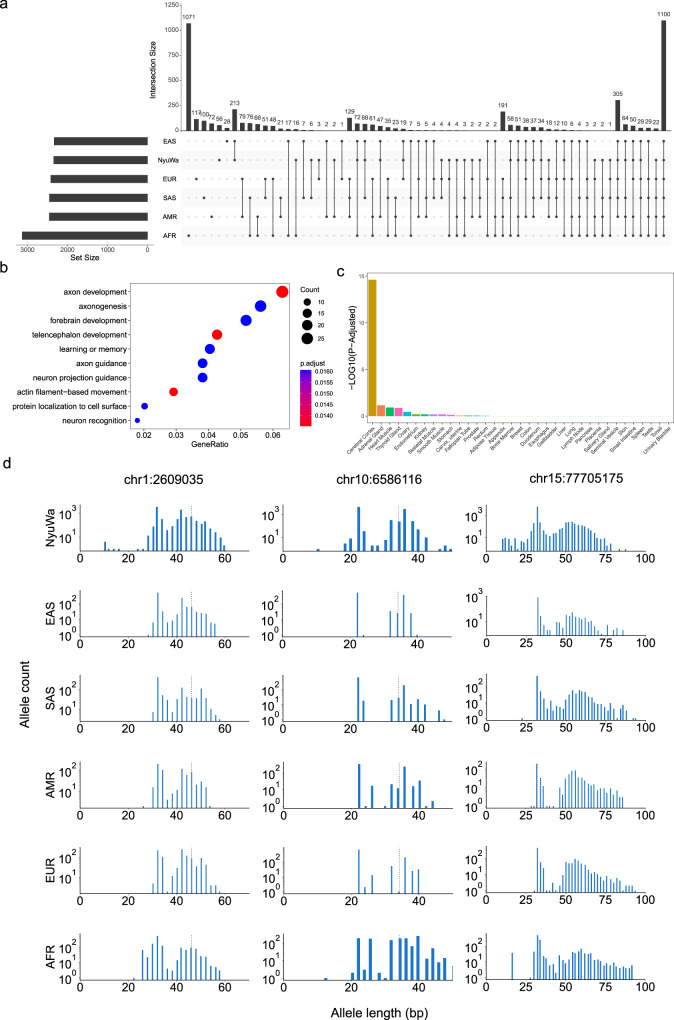
Short tandem repeats (STRs) are abundant and highly mutagenic in the human genome. Many STR loci have been associated with a range of human genetic disorders. However, most population-scale studies on STR variation in humans have focused on European ancestry cohorts or are limited by sequencing depth. Here, we depicted a comprehensive map of 366,013 polymorphic STRs (pSTRs) constructed from 6487 deeply sequenced genomes, comprising 3983 Chinese samples (~31.5x, NyuWa) and 2504 samples from the 1000 Genomes Project (~33.3x, 1KGP). We found that STR mutations were affected by motif length, chromosome context and epigenetic features. We identified 3273 and 1117 pSTRs whose repeat numbers were associated with gene expression and 3'UTR alternative polyadenylation, respectively. We also implemented population analysis, investigated population differentiated signatures, and genotyped 60 known disease-causing STRs. Overall, this study further extends the scale of STR variation in humans and propels our understanding of the semantics of STRs.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
