

Evolutionary characterization of lung adenocarcinoma morphology in TRACERx
- PMID: 37045996
- PMCID: PMC7614478
- DOI: 10.1038/s41591-023-02230-w
Evolutionary characterization of lung adenocarcinoma morphology in TRACERx
Abstract
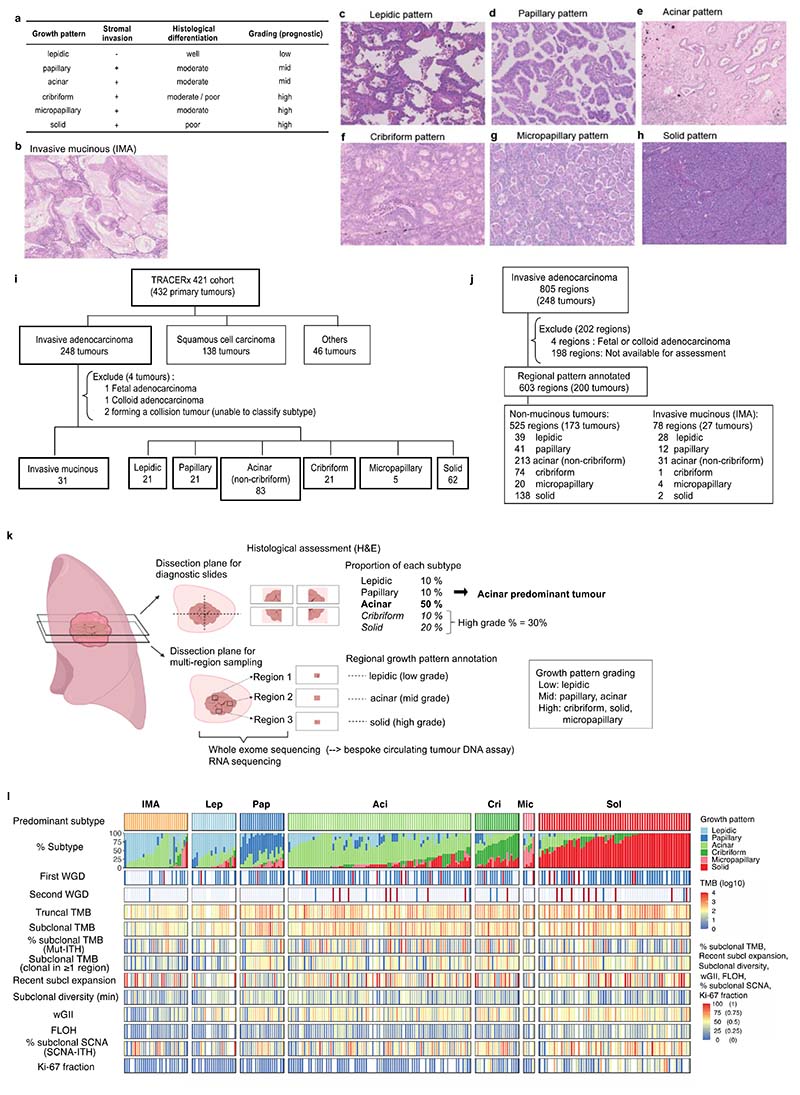
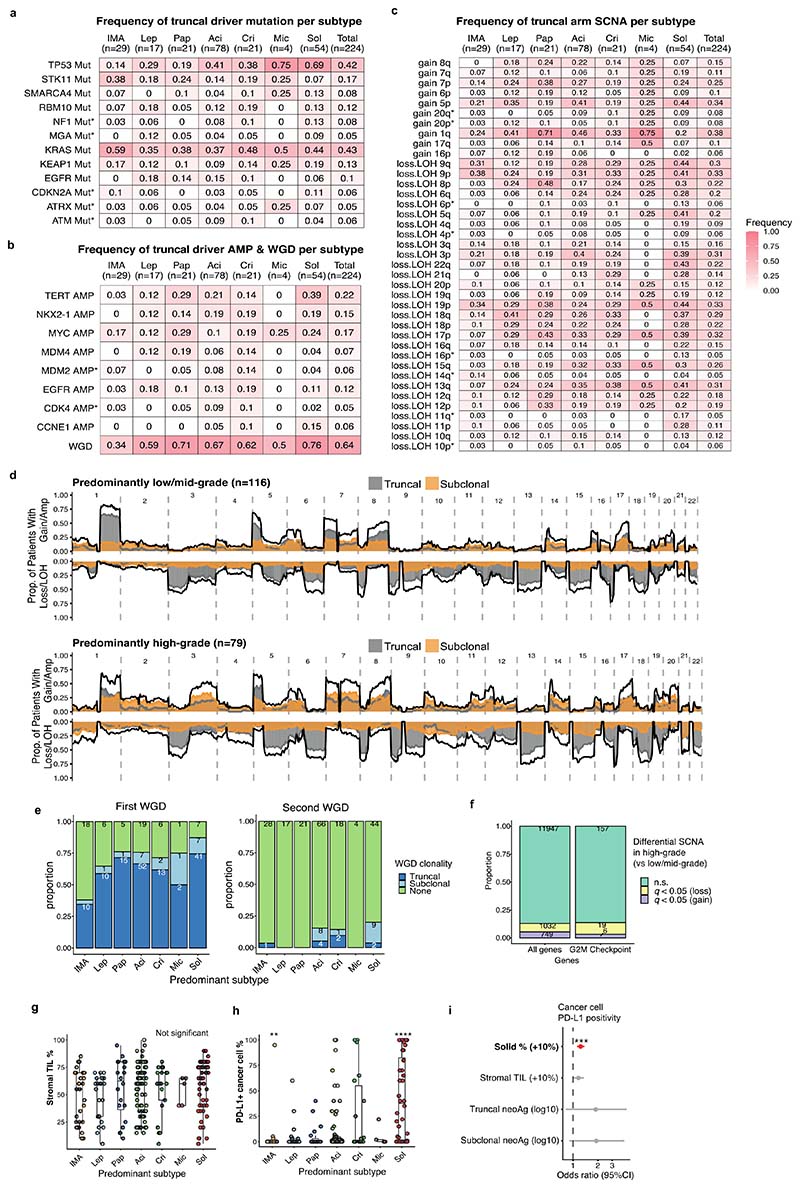
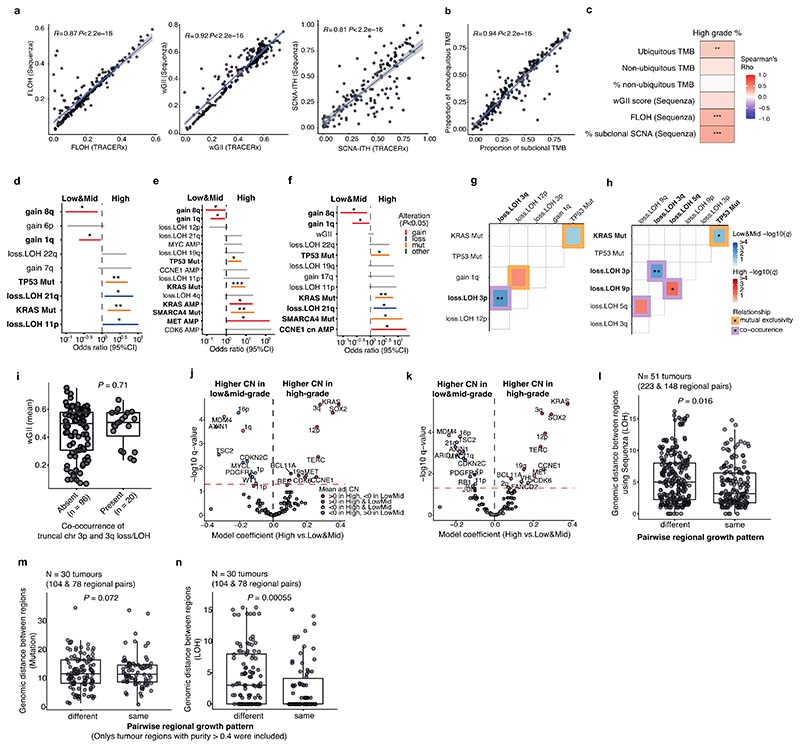
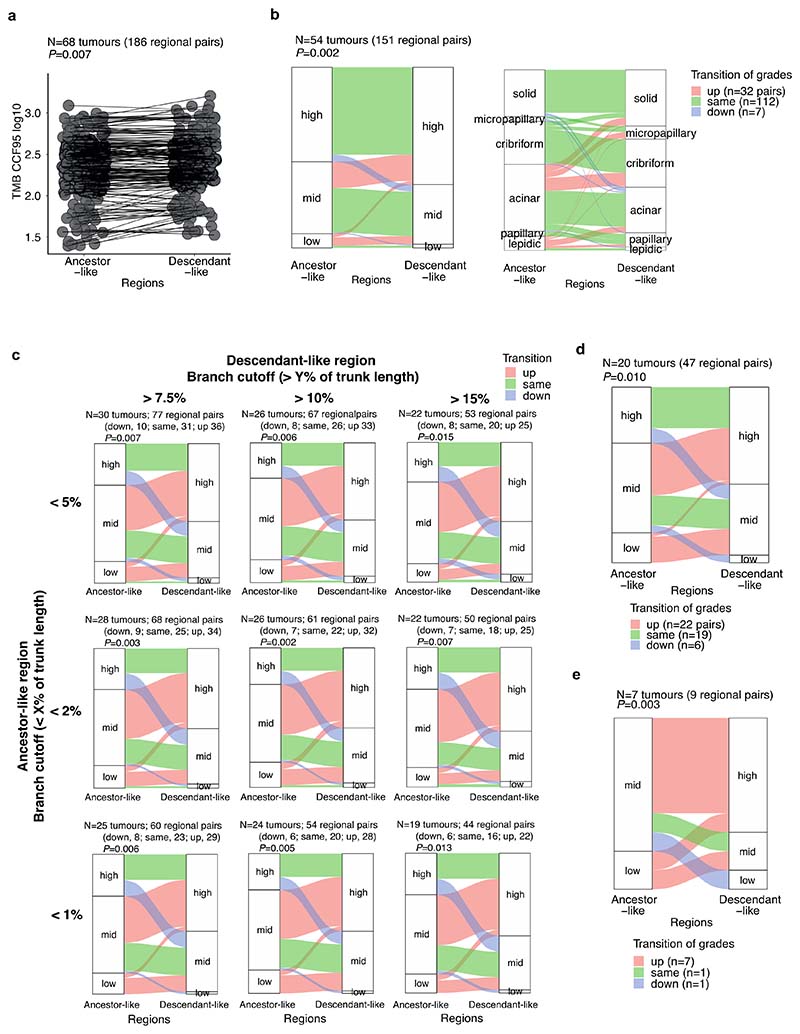
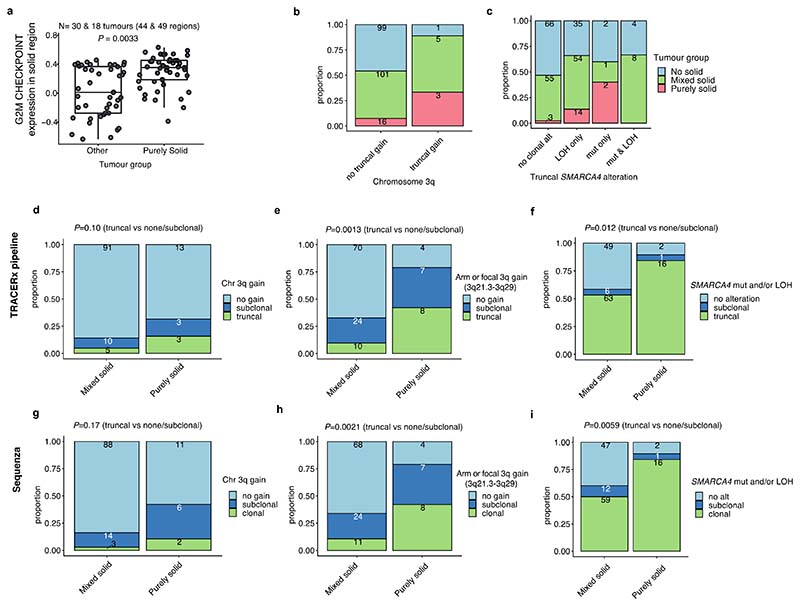
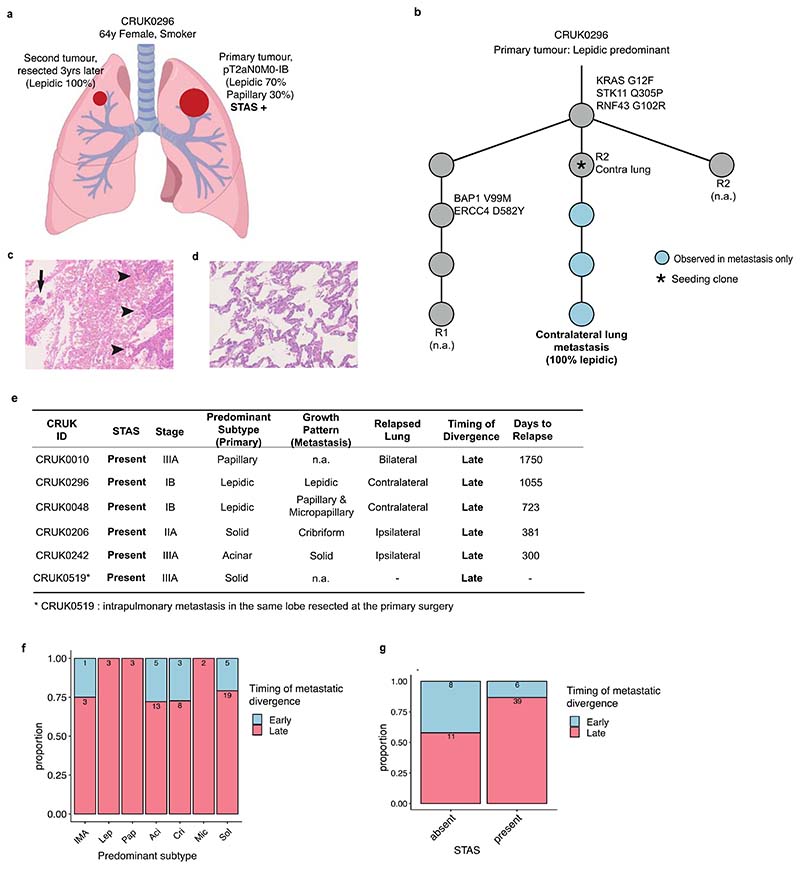
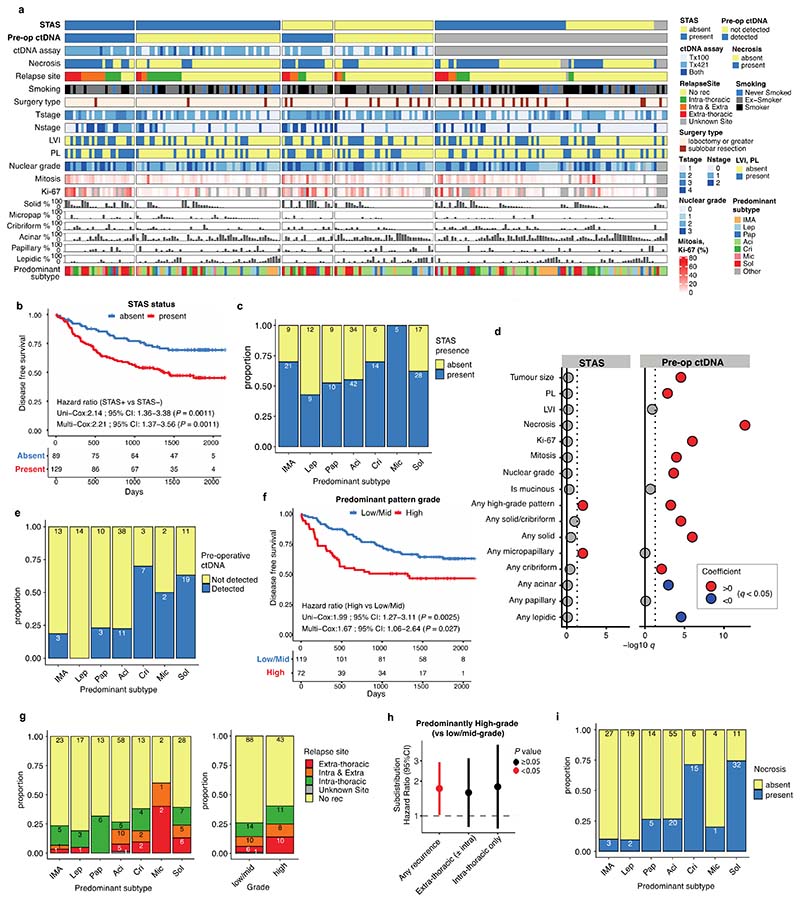
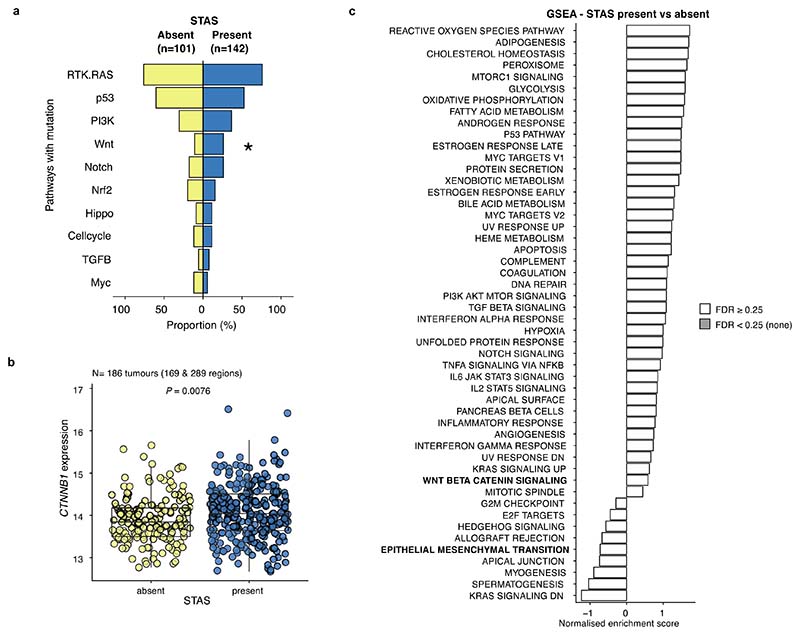
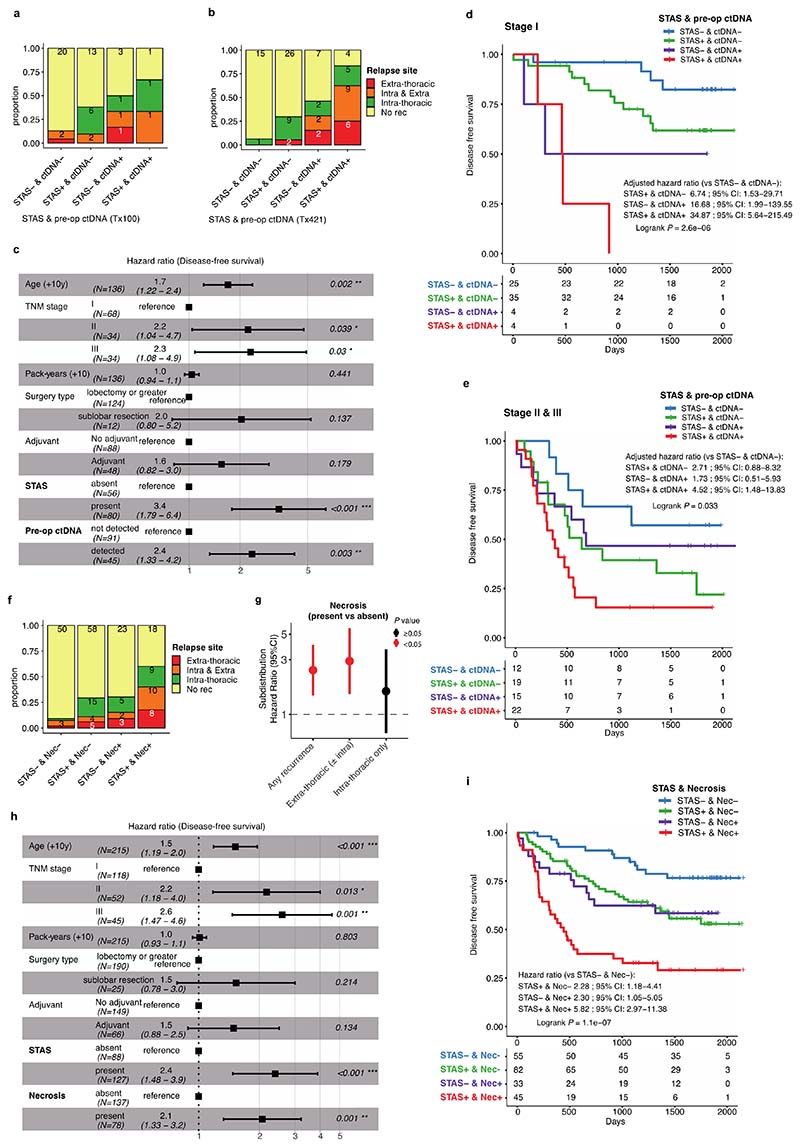
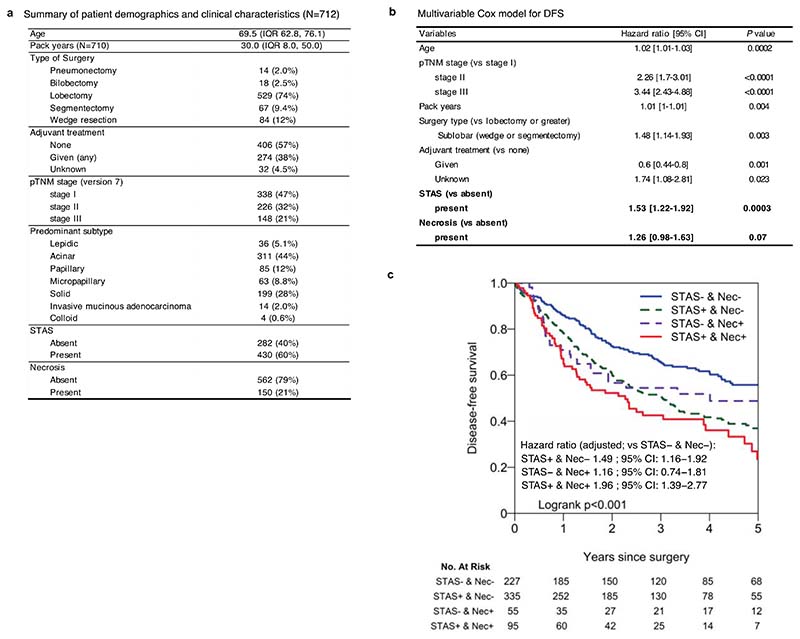
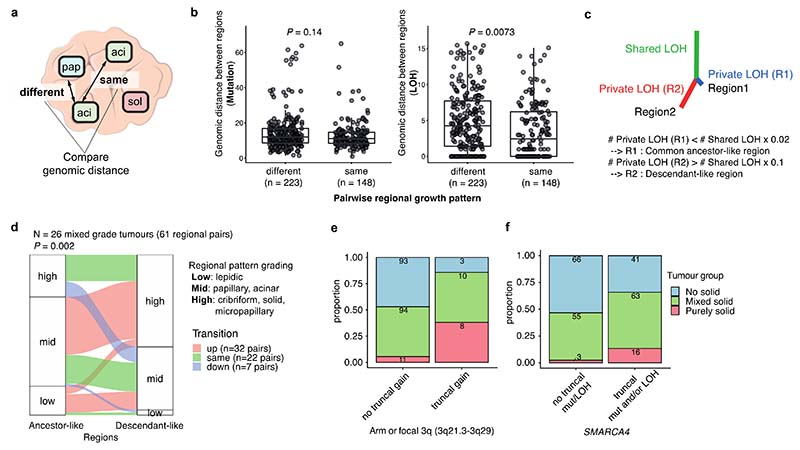
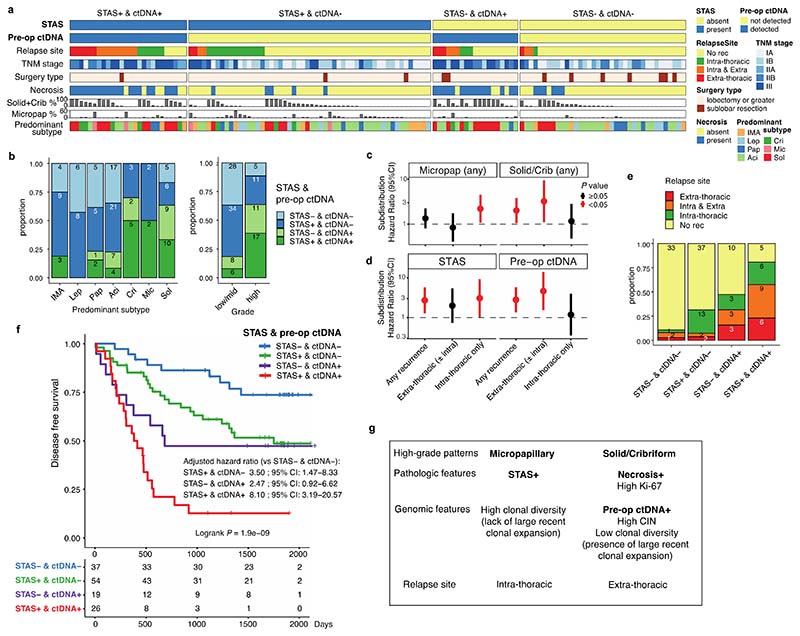
Lung adenocarcinomas (LUADs) display a broad histological spectrum from low-grade lepidic tumors through to mid-grade acinar and papillary and high-grade solid, cribriform and micropapillary tumors. How morphology reflects tumor evolution and disease progression is poorly understood. Whole-exome sequencing data generated from 805 primary tumor regions and 121 paired metastatic samples across 248 LUADs from the TRACERx 421 cohort, together with RNA-sequencing data from 463 primary tumor regions, were integrated with detailed whole-tumor and regional histopathological analysis. Tumors with predominantly high-grade patterns showed increased chromosomal complexity, with higher burden of loss of heterozygosity and subclonal somatic copy number alterations. Individual regions in predominantly high-grade pattern tumors exhibited higher proliferation and lower clonal diversity, potentially reflecting large recent subclonal expansions. Co-occurrence of truncal loss of chromosomes 3p and 3q was enriched in predominantly low-/mid-grade tumors, while purely undifferentiated solid-pattern tumors had a higher frequency of truncal arm or focal 3q gains and SMARCA4 gene alterations compared with mixed-pattern tumors with a solid component, suggesting distinct evolutionary trajectories. Clonal evolution analysis revealed that tumors tend to evolve toward higher-grade patterns. The presence of micropapillary pattern and 'tumor spread through air spaces' were associated with intrathoracic recurrence, in contrast to the presence of solid/cribriform patterns, necrosis and preoperative circulating tumor DNA detection, which were associated with extra-thoracic recurrence. These data provide insights into the relationship between LUAD morphology, the underlying evolutionary genomic landscape, and clinical and anatomical relapse risk.
© 2023. The Author(s) under exclusive license to Springer Nature America, Inc.
Conflict of interest statement
D.A.M. reports speaker fees from AstraZeneca and Takeda, consultancy fees from AstraZeneca, Thermo Fisher, Takeda, Amgen, Janssen and Eli Lilly and has received educational support from Takeda and Amgen. S.V is a co-inventor to a patent to detecting molecules in a sample (Patent # 10578620). M.A.B. has consulted for Achilles Therapeutics D.B. reports personal fees from NanoString and AstraZeneca. He has a patent PCT/GB2020/050221 issued on methods for cancer prognostication. K.S.S.E. received research grant funding from BMS. C.A. has received speaking honoraria or expenses from Novartis, Roche, AstraZeneca and BMS; has patents issued to detect tumour recurrence (PCT/GB2017/053289) and methods for lung cancer detection (PCT/US2017/028013); and is a current employee of AstraZeneca. C.T.H. has received speaker fees from AstraZeneca. K.L. has a patent on indel burden and CPI response pending and speaker fees from Roche tissue diagnostics, research funding from CRUK TDL/Ono/LifeArc alliance, and a consulting role with Monopteros Therapeutics. N.J.B. is a co-inventor to a patent to identifying responders to cancer treatment (PCT/GB2018/051912). E.L.C. is employed by and holds shares of Achilles Therapeutics. P.S.A. is a Scientific Advisory Board Member and Consultant for ATARA Biotherapeutics, Bayer, Carisma Therapeutics, Imugene, ImmPactBio, Johnston & Johnston, Orion, OutpaceBio; declares patents, royalties and intellectual property on mesothelin-targeted CAR and other T-cell therapies, which have been licensed to ATARA Biotherapeutics, issued patent method for detection of cancer cells using virus, and pending patent applications on PD-1 dominant negative receptor, wireless pulse-oximetry device, and on an ex vivo malignant pleural effusion culture system. D.R.J. has consulted for AstraZeneca and is a member of Clinical Trial Steering Committee for Merck. N.R. serves on an NCI Thoracic Malignancies Steering Committee (which is a compensated role). W.D.T. is a non-paid consultant for the LCMC3 and LCMC4 neoadjuvant clinical trials. A.Ha. has received fees for member of Independent Data Monitoring Committees for Roche-sponsored clinical trials, and academic projects co-ordinated by Roche. R.S. reports research funding by Roche, Merck, and Puma Biotechnology; travel and congress-registration support by Roche, Merck, and Astra Zeneca; and was part of Advisory Boards for Bristol Myers Squibb and Roche. A.G.N reports personal fees from Merck, Boehringer Ingelheim, Novartis, Astra Zeneca, Bristol Myer Squib, Roche, Abbvie, Oncologica, Uptodate, European Society of Oncology, Takeda UK, and Liberium, and personal fees and grants from Pfizer. N.M. has stock options in and has consulted for Achilles Therapeutics and holds a European patent in determining HLA LOH (PCT/GB2018/052004), and is a co-inventor to a patent to identifying responders to cancer treatment (PCT/GB2018/051912). M.J-H is a CRUK Career Establishment Awardee and has received funding from CRUK, IASLC International Lung Cancer Foundation, Lung Cancer Research Foundation, Rosetrees Trust, UKI NETs, NIHR, NIHR UCLH Biomedical Research Centre. M.J-H. has consulted, and is a member of the Scientific Advisory Board and Steering Committee, for Achilles Therapeutics, has received speaker honoraria from Astex Pharmaceuticals, Oslo Cancer Cluster, and holds a patent PCT/US2017/028013 relating to methods for lung cancer detection. C.S. acknowledges grant support from Pfizer, AstraZeneca, Bristol Myers Squibb, Roche-Ventana, Boehringer-Ingelheim, Archer Dx Inc. (collaboration in minimal residual disease sequencing technologies) and Ono Pharmaceutical; is an AstraZeneca Advisory Board Member and Chief Investigator for the MeRmaiD1 clinical trial; has consulted for Amgen, Pfizer, Novartis, GlaxoSmithKline, MSD, Bristol Myers Squibb, AstraZeneca, Illumina, Genentech, Roche-Ventana, GRAIL, Medicxi, Bicycle Therapeutics, Metabomed and the Sarah Cannon Research Institute; has stock options in Apogen Biotechnologies, Epic Bioscience and GRAIL; and has stock options and is co-founder of Achilles Therapeutics. C.S. holds patents relating to assay technology to detect tumour recurrence (PCT/GB2017/053289); to targeting neoantigens (PCT/EP2016/059401), identifying patent response to immune checkpoint blockade (PCT/EP2016/071471), determining HLA LOH (PCT/GB2018/052004), predicting survival rates of patients with cancer (PCT/GB2020/050221), to treating cancer by targeting Insertion/deletion mutations (PCT/GB2018/051893); identifying insertion/deletion mutation targets (PCT/GB2018/051892); methods for lung cancer detection (PCT/US2017/028013); and identifying responders to cancer treatment (PCT/GB2018/051912). A.M.F., C.A. and C.S. are named inventors on a patent application to determine methods and systems for tumour monitoring (GB2114434.0).
Figures


Comment in
-
Clinicogenomic landscape of morphological evolution in lung adenocarcinoma.Nat Med. 2023 Apr;29(4):795-796. doi: 10.1038/s41591-023-02231-9. Nat Med. 2023. PMID: 37045998 No abstract available.
References
-
- Nicholson AG, et al. The 2021 WHO Classification of Lung Tumors: Impact of Advances Since 2015. J Thorac Oncol. 2022;17:362–387. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- 211179/Z/18/Z/WT_/Wellcome Trust/United Kingdom
- FC001202/WT_/Wellcome Trust/United Kingdom
- 21999/CRUK_/Cancer Research UK/United Kingdom
- CTRNBC-2022/100001/CRUK_/Cancer Research UK/United Kingdom
- C11496/A17786/CRUK_/Cancer Research UK/United Kingdom
- 24956/CRUK_/Cancer Research UK/United Kingdom
- R01 CA235667/CA/NCI NIH HHS/United States
- 17786/CRUK_/Cancer Research UK/United Kingdom
- MR/V033077/1/MRC_/Medical Research Council/United Kingdom
- 30025/CRUK_/Cancer Research UK/United Kingdom
- C416/A21999/CRUK_/Cancer Research UK/United Kingdom
- P30 CA008748/CA/NCI NIH HHS/United States
- R01 CA236615/CA/NCI NIH HHS/United States
- MR/P014712/1 /MRC_/Medical Research Council/United Kingdom
- R01 CA240472/CA/NCI NIH HHS/United States
- C11496/A30025/CRUK_/Cancer Research UK/United Kingdom
- 29569/CRUK_/Cancer Research UK/United Kingdom
- CTUQQR-DEC22/100009/CRUK_/Cancer Research UK/United Kingdom
- C444/A15953/CRUK_/Cancer Research UK/United Kingdom
- FC001202/CRUK_/Cancer Research UK/United Kingdom
- FC001169/WT_/Wellcome Trust/United Kingdom
- C69256/A30194/CRUK_/Cancer Research UK/United Kingdom
- C7893/A26233/CRUK_/Cancer Research UK/United Kingdom
- CC2008/MRC_/Medical Research Council/United Kingdom
- U01 CA214195/CA/NCI NIH HHS/United States
- CC2041/MRC_/Medical Research Council/United Kingdom
- R01 CA217169/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
