

Aberrant activation of TCL1A promotes stem cell expansion in clonal haematopoiesis
- PMID: 37046083
- PMCID: PMC10360040
- DOI: 10.1038/s41586-023-05806-1
Aberrant activation of TCL1A promotes stem cell expansion in clonal haematopoiesis
Abstract
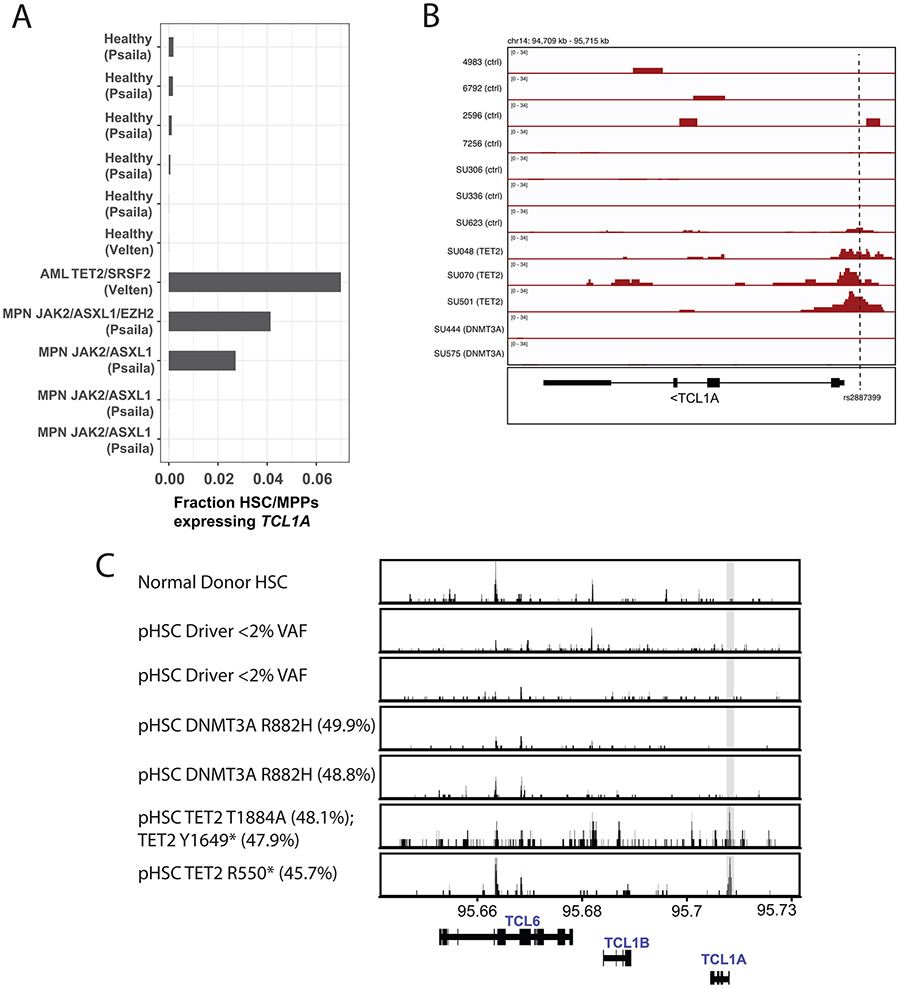
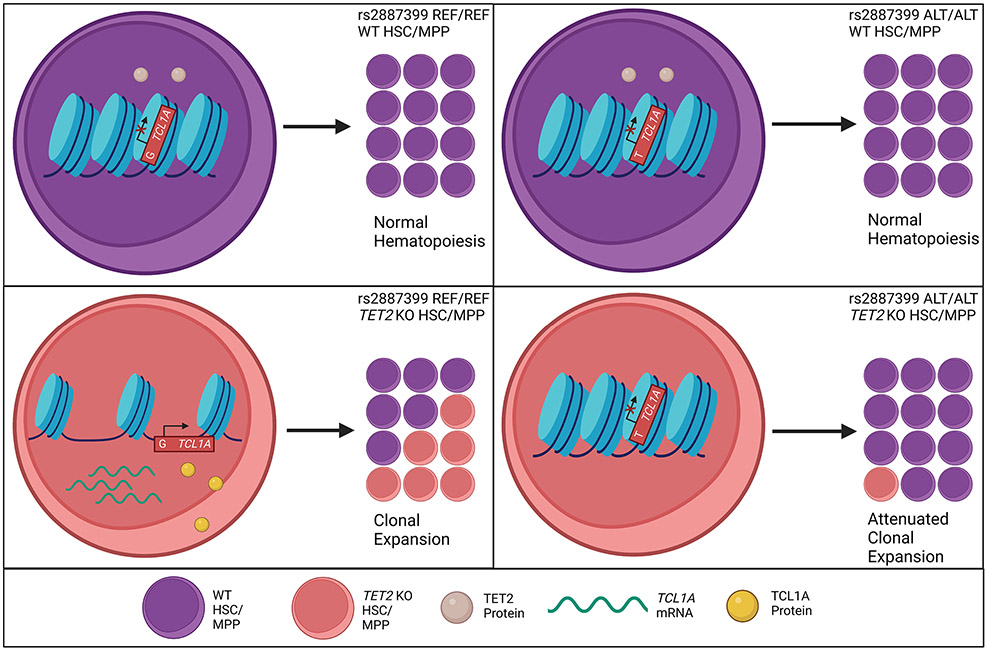
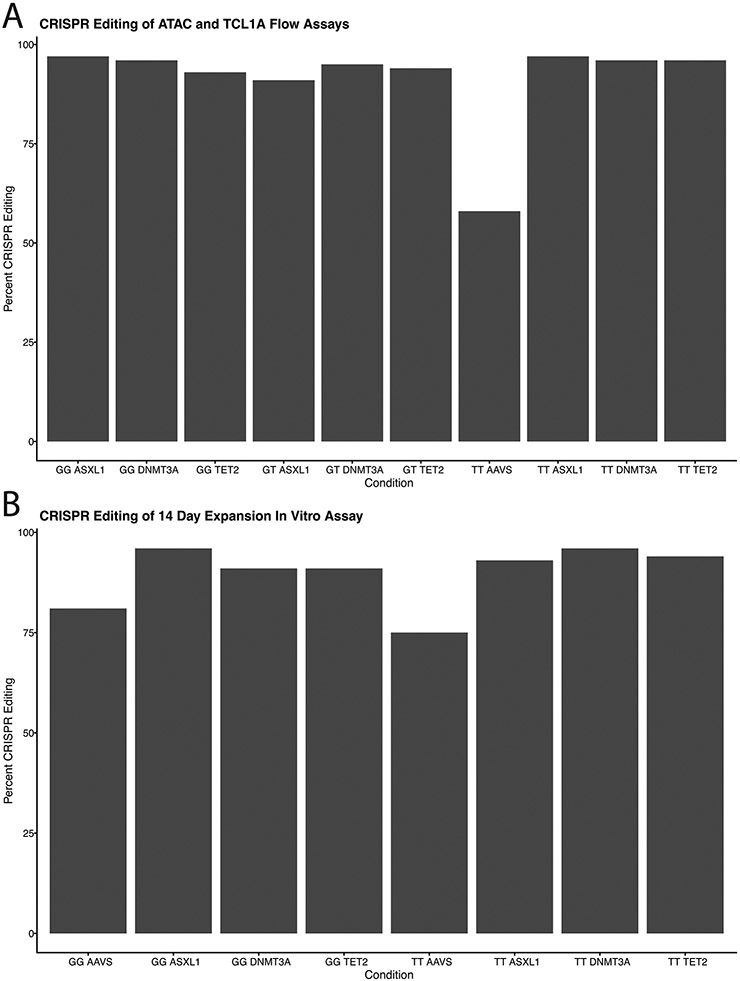
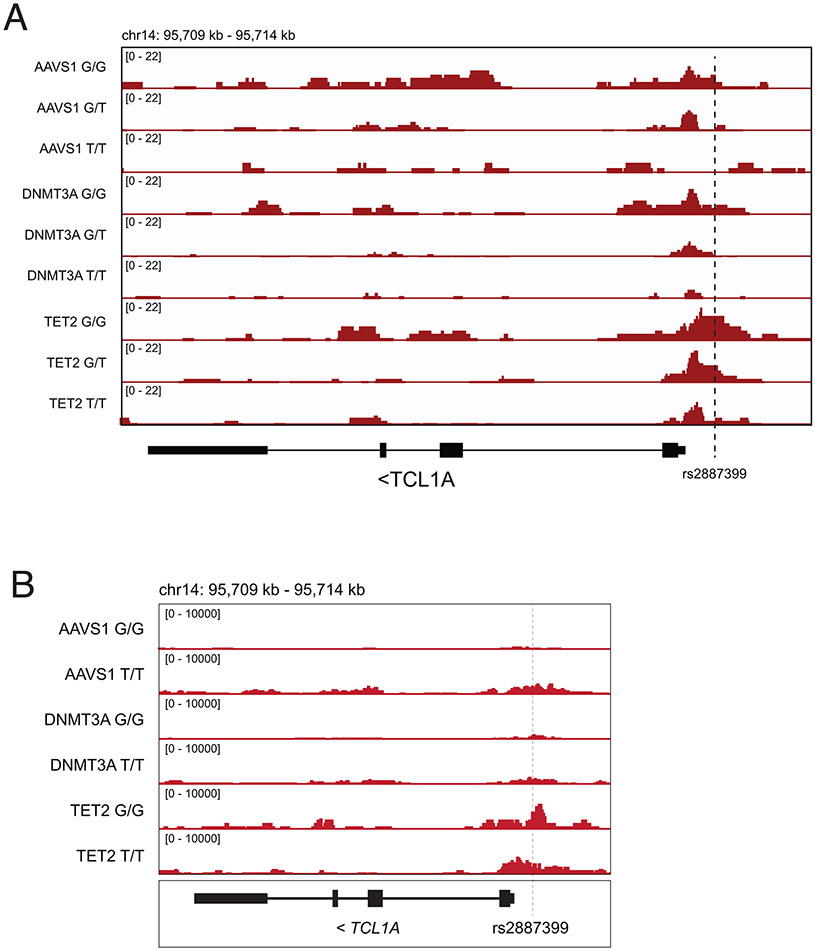
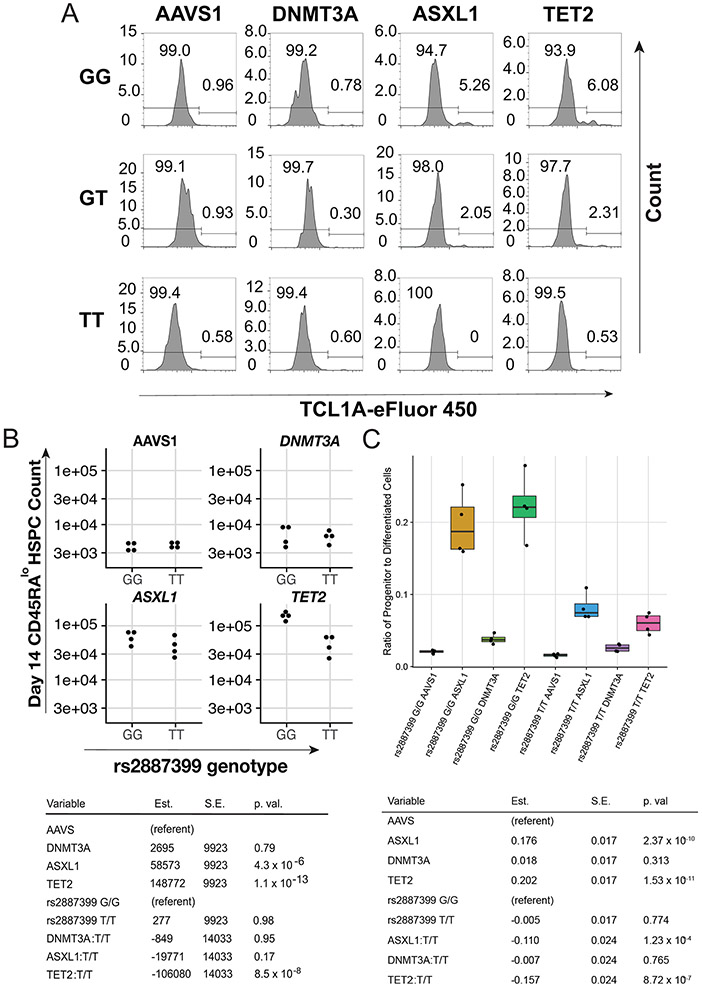
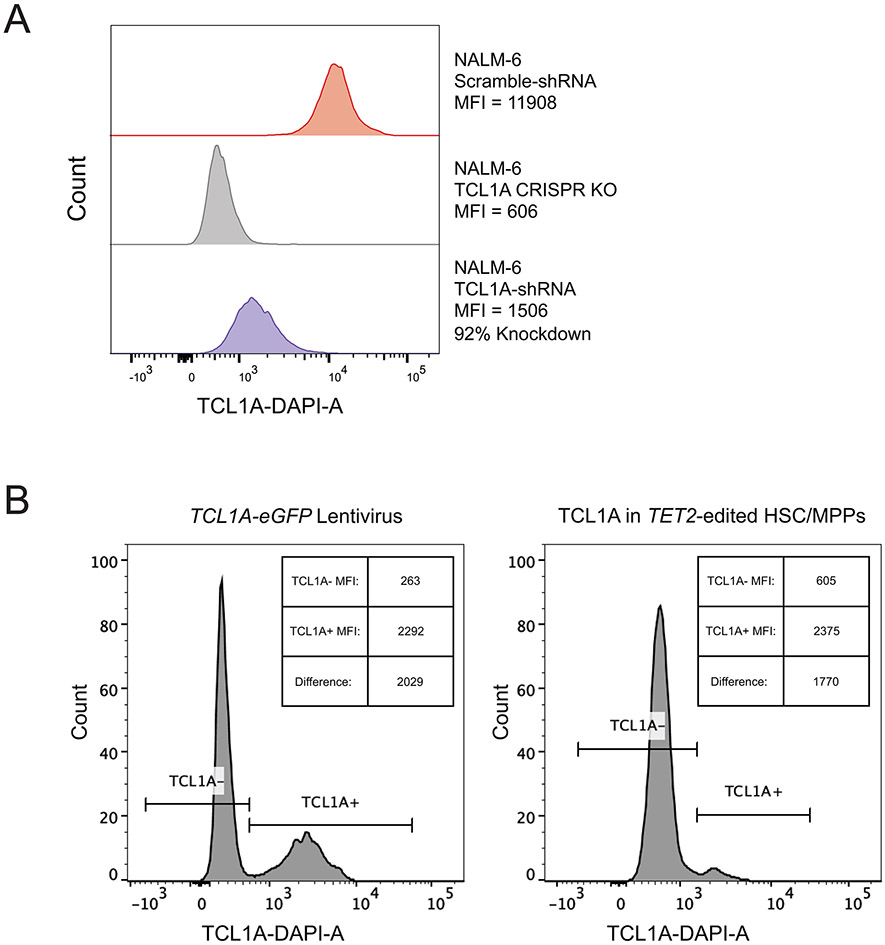
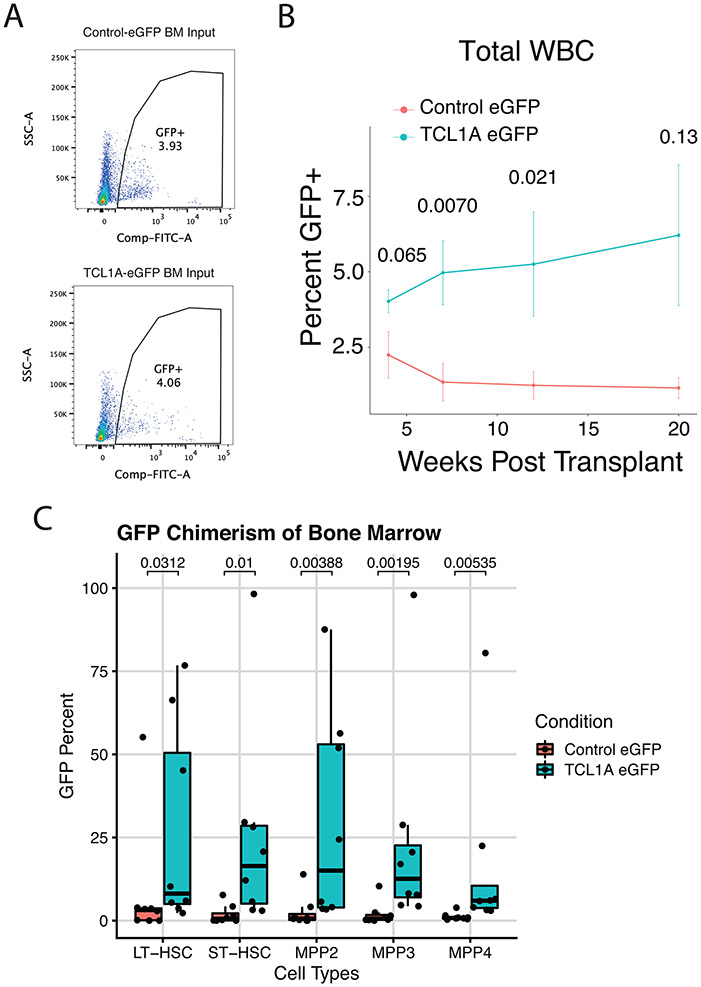
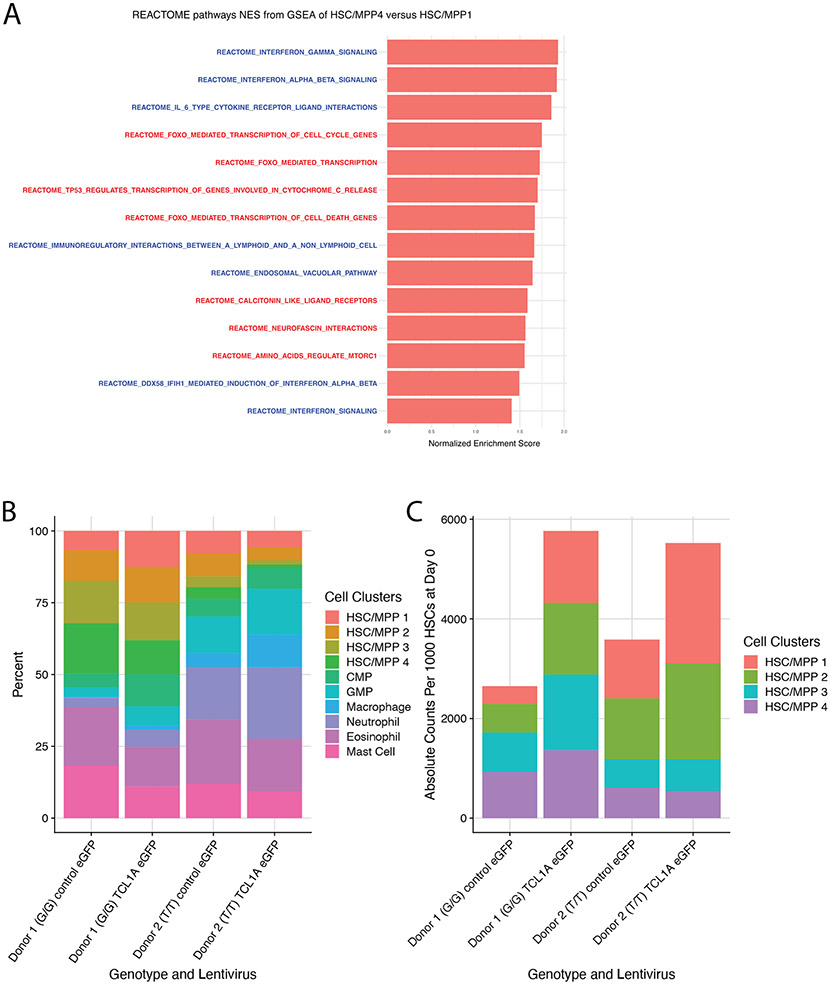
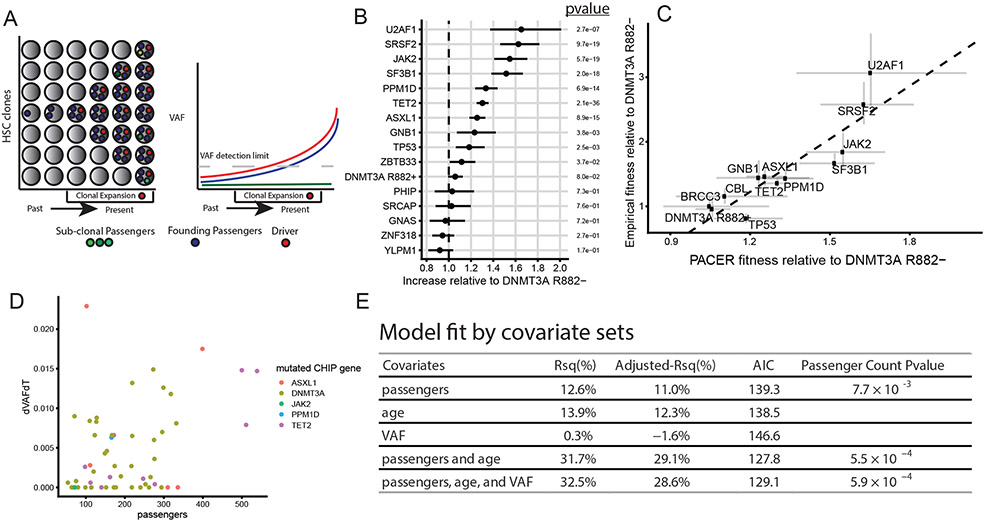
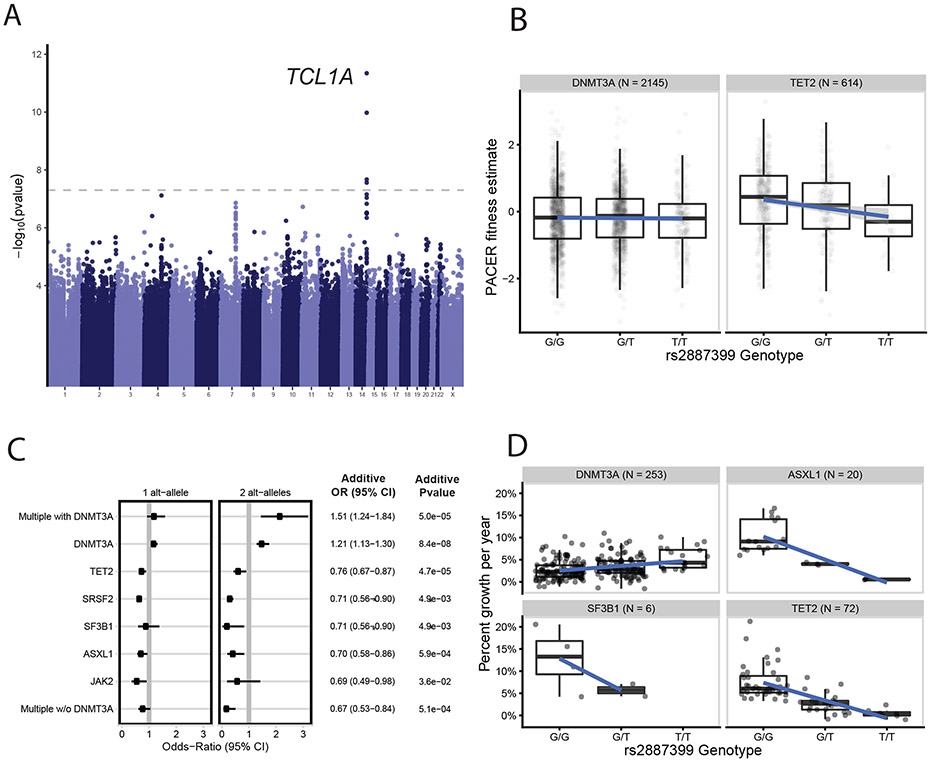
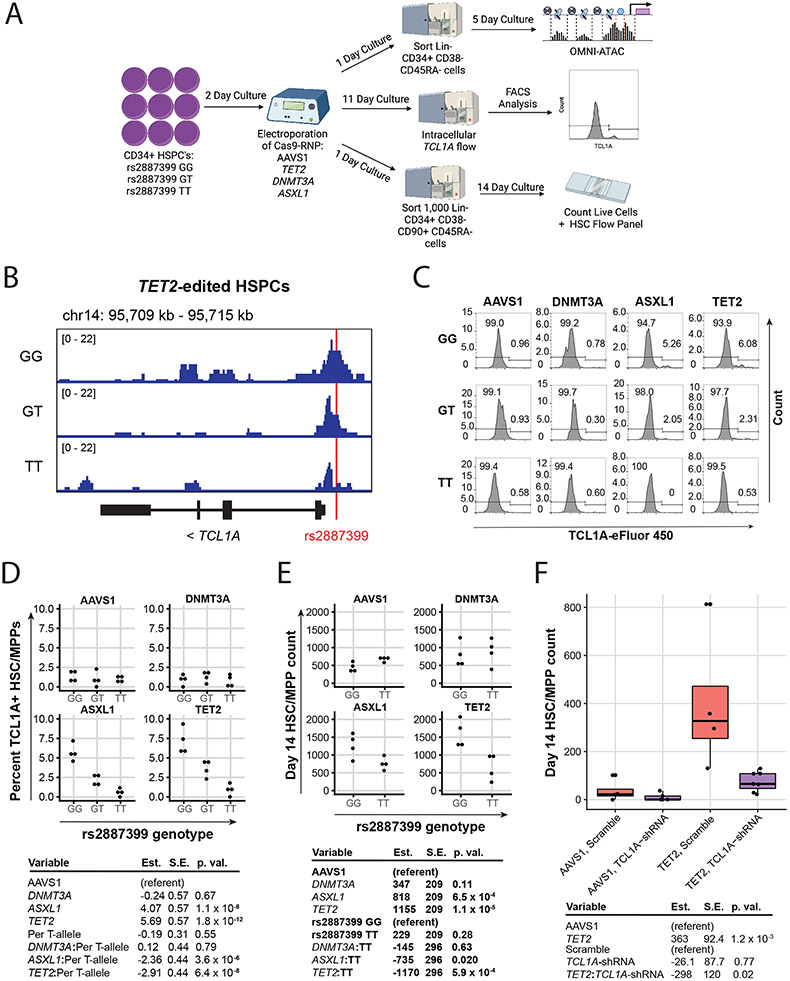
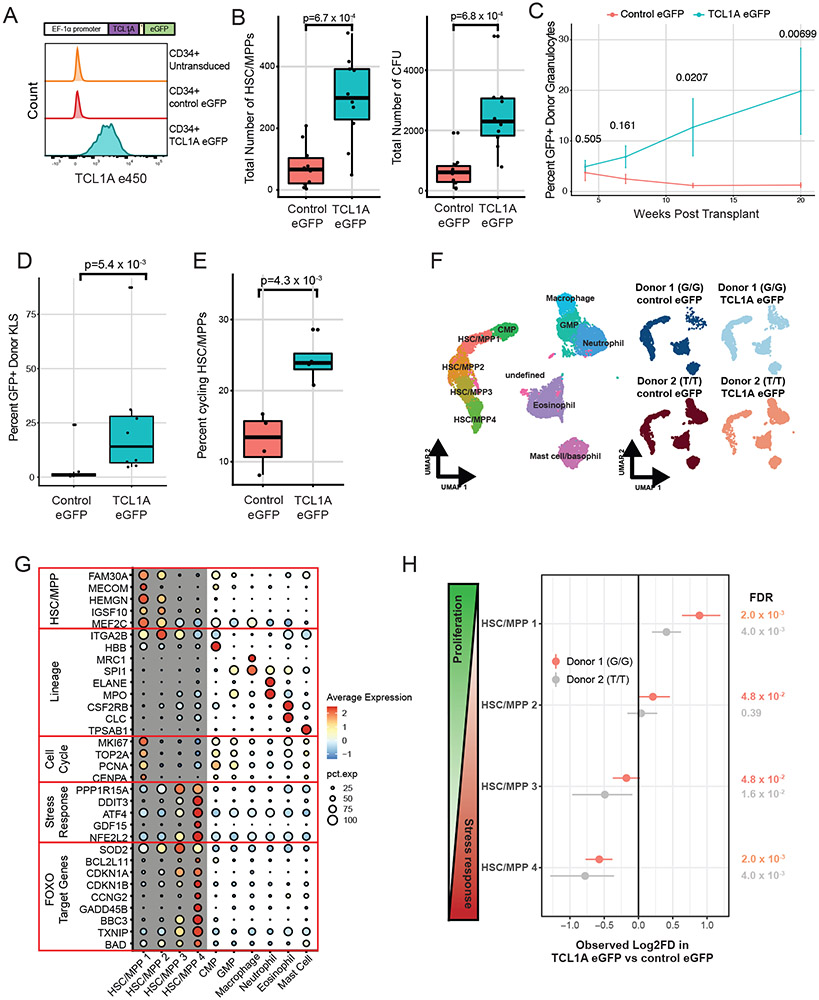
Mutations in a diverse set of driver genes increase the fitness of haematopoietic stem cells (HSCs), leading to clonal haematopoiesis1. These lesions are precursors for blood cancers2-6, but the basis of their fitness advantage remains largely unknown, partly owing to a paucity of large cohorts in which the clonal expansion rate has been assessed by longitudinal sampling. Here, to circumvent this limitation, we developed a method to infer the expansion rate from data from a single time point. We applied this method to 5,071 people with clonal haematopoiesis. A genome-wide association study revealed that a common inherited polymorphism in the TCL1A promoter was associated with a slower expansion rate in clonal haematopoiesis overall, but the effect varied by driver gene. Those carrying this protective allele exhibited markedly reduced growth rates or prevalence of clones with driver mutations in TET2, ASXL1, SF3B1 and SRSF2, but this effect was not seen in clones with driver mutations in DNMT3A. TCL1A was not expressed in normal or DNMT3A-mutated HSCs, but the introduction of mutations in TET2 or ASXL1 led to the expression of TCL1A protein and the expansion of HSCs in vitro. The protective allele restricted TCL1A expression and expansion of mutant HSCs, as did experimental knockdown of TCL1A expression. Forced expression of TCL1A promoted the expansion of human HSCs in vitro and mouse HSCs in vivo. Our results indicate that the fitness advantage of several commonly mutated driver genes in clonal haematopoiesis may be mediated by TCL1A activation.
© 2023. The Author(s), under exclusive licence to Springer Nature Limited.
Figures



Comment in
-
Linking TCL1A to clonal expansions in blood.Nat Genet. 2023 May;55(5):727. doi: 10.1038/s41588-023-01406-x. Nat Genet. 2023. PMID: 37173527 No abstract available.
-
Pacing of clonal expansion is dictated by its underlying mutation.Nat Cardiovasc Res. 2024 Apr;3(4):404. doi: 10.1038/s44161-024-00467-3. Nat Cardiovasc Res. 2024. PMID: 39196221 No abstract available.
References
WORKS CITED MAIN TEXT
WORKS CITED METHODS
-
- Voss K, Gentry J & Van der Auwera G Full-stack genomics pipelining with GATK4 + WDL + Cromwell. in (F1000 Research, 2017). doi: 10.7490/f1000research.1114631.1. - DOI
Publication types
MeSH terms
Substances
Grants and funding
- P30 DK040561/DK/NIDDK NIH HHS/United States
- R01 HL148565/HL/NHLBI NIH HHS/United States
- P50 HL118006/HL/NHLBI NIH HHS/United States
- S10 OD026880/OD/NIH HHS/United States
- T32 HL098049/HL/NHLBI NIH HHS/United States
- HHSN268201800001C/HL/NHLBI NIH HHS/United States
- R01 AI132476/AI/NIAID NIH HHS/United States
- DP2 HL157540/HL/NHLBI NIH HHS/United States
- S10 OD030463/OD/NIH HHS/United States
- R01 HL143818/HL/NHLBI NIH HHS/United States
- U54 DK106829/DK/NIDDK NIH HHS/United States
- HHSN268201000001I/HL/NHLBI NIH HHS/United States
- DP5 OD029586/OD/NIH HHS/United States
- R01 HL120393/HL/NHLBI NIH HHS/United States
- R01 HL125005/HL/NHLBI NIH HHS/United States
- K01 AG059898/AG/NIA NIH HHS/United States
- U01 HL120393/HL/NHLBI NIH HHS/United States
- R01 HL153805/HL/NHLBI NIH HHS/United States
- R01 HL117626/HL/NHLBI NIH HHS/United States
- K08 HL146972/HL/NHLBI NIH HHS/United States
- P01 HL132825/HL/NHLBI NIH HHS/United States
- R01 HL148050/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
