

Genomic-transcriptomic evolution in lung cancer and metastasis
- PMID: 37046093
- PMCID: PMC10115639
- DOI: 10.1038/s41586-023-05706-4
Genomic-transcriptomic evolution in lung cancer and metastasis
Abstract
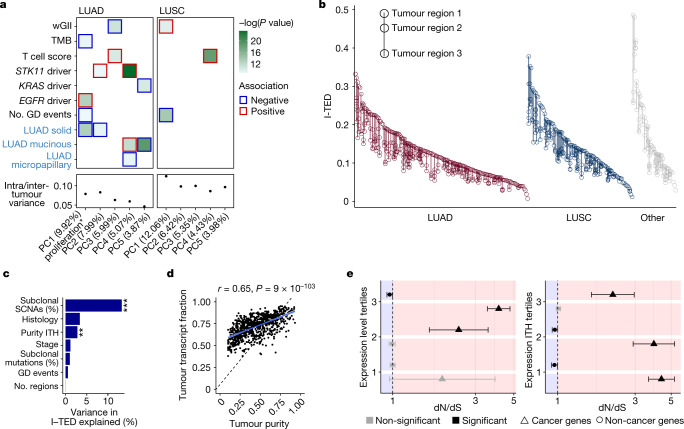
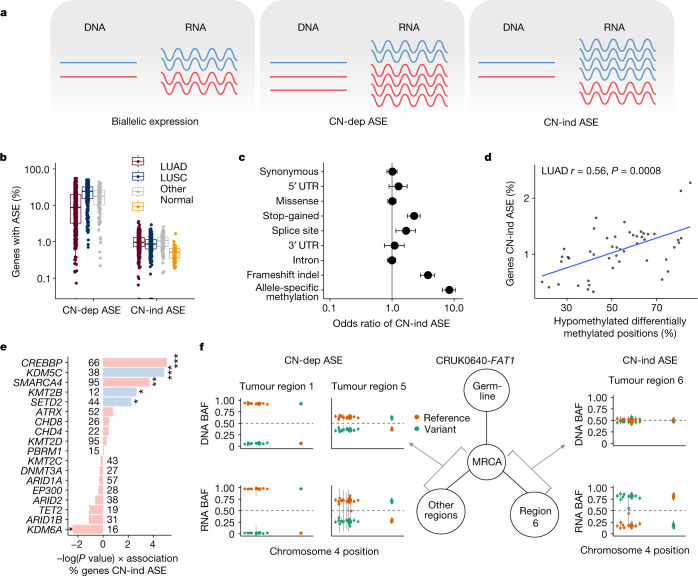
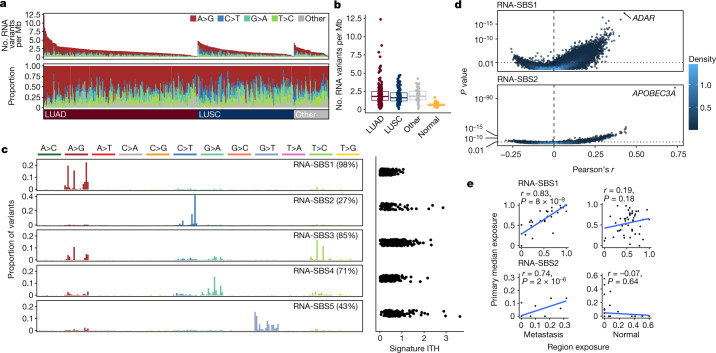
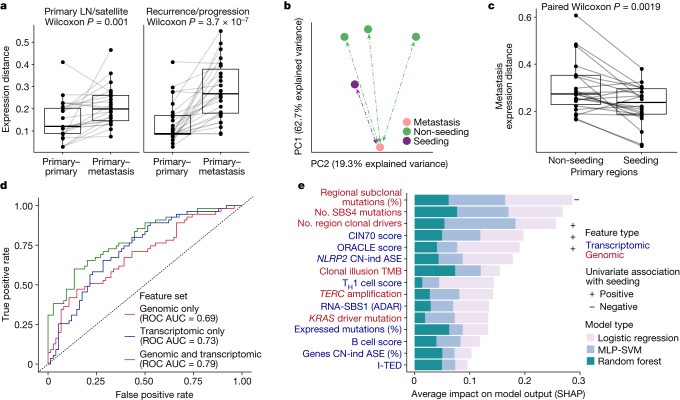
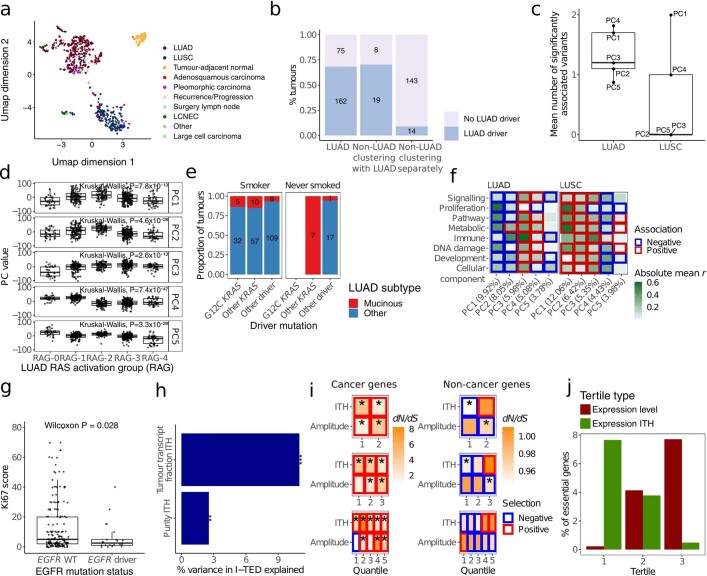
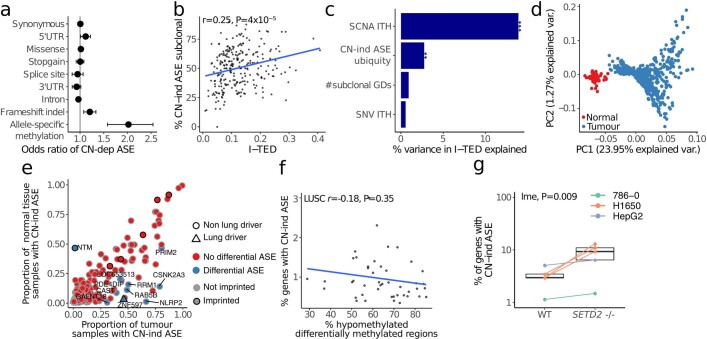
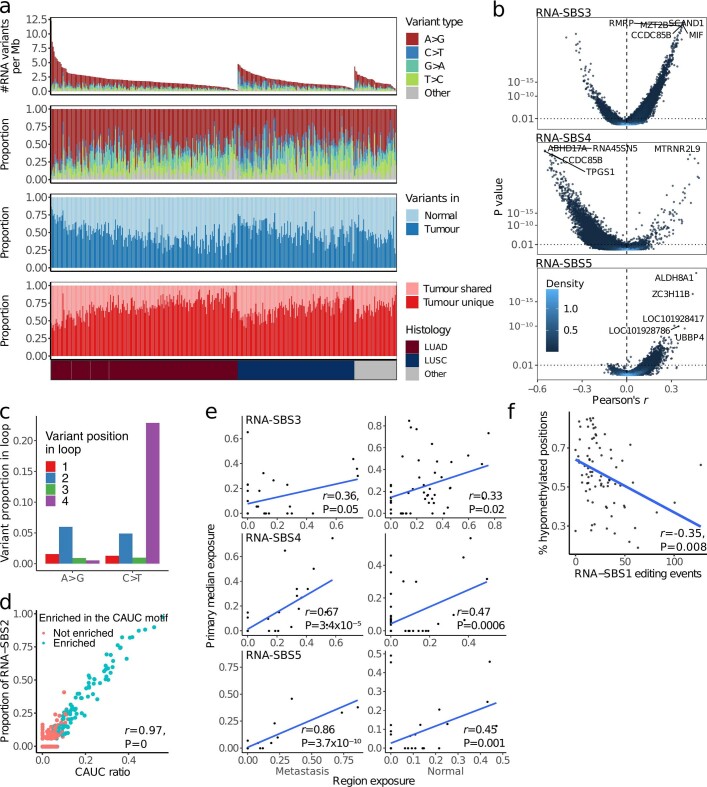
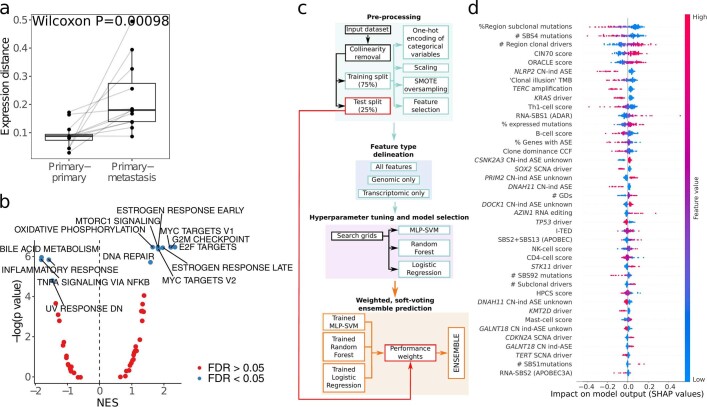
Intratumour heterogeneity (ITH) fuels lung cancer evolution, which leads to immune evasion and resistance to therapy1. Here, using paired whole-exome and RNA sequencing data, we investigate intratumour transcriptomic diversity in 354 non-small cell lung cancer tumours from 347 out of the first 421 patients prospectively recruited into the TRACERx study2,3. Analyses of 947 tumour regions, representing both primary and metastatic disease, alongside 96 tumour-adjacent normal tissue samples implicate the transcriptome as a major source of phenotypic variation. Gene expression levels and ITH relate to patterns of positive and negative selection during tumour evolution. We observe frequent copy number-independent allele-specific expression that is linked to epigenomic dysfunction. Allele-specific expression can also result in genomic-transcriptomic parallel evolution, which converges on cancer gene disruption. We extract signatures of RNA single-base substitutions and link their aetiology to the activity of the RNA-editing enzymes ADAR and APOBEC3A, thereby revealing otherwise undetected ongoing APOBEC activity in tumours. Characterizing the transcriptomes of primary-metastatic tumour pairs, we combine multiple machine-learning approaches that leverage genomic and transcriptomic variables to link metastasis-seeding potential to the evolutionary context of mutations and increased proliferation within primary tumour regions. These results highlight the interplay between the genome and transcriptome in influencing ITH, lung cancer evolution and metastasis.
Trial registration: ClinicalTrials.gov NCT01888601.
© 2023. The Author(s).
Conflict of interest statement
S.V. is a co-inventor to a patent to detect molecules in a sample (US patent no. 10578620). M.A.B. has consulted for Achilles Therapeutics. A.M.F. is co-inventor to a patent application to determine methods and systems for tumour monitoring (PCT/EP2022/077987). M.J-H. has consulted for, and is a member of, the Achilles Therapeutics Scientific Advisory Board and Steering Committee; has received speaker honoraria from Pfizer, Astex Pharmaceuticals and Oslo Cancer Cluster; and holds patent PCT/US2017/028013 relating to methods for lung cancer detection. This patent has been licensed to commercial entities and under terms of employment. M.J.-H. is due a share of any revenue generated from such license(s). A. Hackshaw has received fees for being a member of independent data monitoring committees for Roche-sponsored clinical trials and academic projects co-ordinated by Roche. C.S. acknowledges grant support from AstraZeneca, Boehringer-Ingelheim, Bristol Myers Squibb, Pfizer, Roche-Ventana, Invitae (previously Archer Dx Inc — collaboration in minimal residual disease sequencing technologies) and Ono Pharmaceutical. C.S. is an AstraZeneca Advisory Board member and chief investigator for the AZ MeRmaiD 1 and 2 clinical trials, and is also co-chief investigator of the NHS Galleri trial, funded by GRAIL, and a paid member of GRAIL’s Scientific Advisory Board. He receives consultant fees from Achilles Therapeutics (where he is also a Scientific Advisory Board member), Bicycle Therapeutics (where he is also a Scientific Advisory Board member), Genentech, Medicxi, Roche Innovation Centre – Shanghai, Metabomed (until July 2022) and the Sarah Cannon Research Institute, had stock options in Apogen Biotechnologies and GRAIL until June 2021, currently has stock options in Epic Bioscience and Bicycle Therapeutics, and has stock options in and is co-founder of Achilles Therapeutics. C.S. is an inventor on a European patent application relating to assay technology to detect tumour recurrence (PCT/GB2017/053289); the patent has been licensed to commercial entities and under his terms of employment C.S is due a revenue share of any revenue generated from such license(s). C.S. holds patents relating to targeting neoantigens (PCT/EP2016/059401), identifying clinical response to immune checkpoint blockade (PCT/EP2016/071471), determining HLA loss of heterozygosity (PCT/GB2018/052004), predicting survival rates of patients with cancer (PCT/GB2020/050221), identifying patients whose cancer responds to treatment (PCT/GB2018/051912), detecting tumour mutations (PCT/US2017/28013), methods for lung cancer detection (US20190106751A1) and identifying insertion/deletion mutation targets (European and US, PCT/GB2018/051892), and is co-inventor to a patent application to determine methods and systems for tumour monitoring (PCT/EP2022/077987). C.S. is a named inventor on a provisional patent protection related to a ctDNA detection algorithm. G.A.W. is employed by and has stock options in Achilles Therapeutics. N.M. has received consultancy fees and has stock options in Achilles Therapeutics. N.M. holds European patents relating to targeting neoantigens (PCT/EP2016/ 059401), identifying clinical response to immune checkpoint blockade (PCT/ EP2016/071471), determining HLA loss of heterozygosity (PCT/GB2018/052004) and predicting survival rates of patients with cancer (PCT/GB2020/050221). D.A.M. reports speaker fees from AstraZeneca, Eli Lilly and Takeda, consultancy fees from AstraZeneca, Thermo Fisher, Takeda, Amgen, Janssen and Eli Lilly and has received educational support from Takeda and Amgen. S.C.T. has acted as a consultant for Revolution Medicines. N.J.B. is a co-inventor to a patent to identify patients whose cancer responds to treatment (PCT/GB2018/051912), a co-inventor on a patent for methods for predicting anti-cancer response (US14/466,208) and has a patent application (PCT/GB2020/050221) on methods for cancer prognostication. R.S. reports non-financial support from Merck and Bristol Myers Squibb (BMS); research support from Merck, Puma Biotechnology and Roche; and personal fees from Roche, BMS and Exact Sciences for advisory boards. E.L.C. is employed by and has stocks in Achilles Therapeutics.
Figures
Comment in
-
Molecular portraits of lung cancer evolution.Nature. 2023 Apr;616(7957):435-436. doi: 10.1038/d41586-023-00934-0. Nature. 2023. PMID: 37045956 No abstract available.
References
Publication types
MeSH terms
Substances
Associated data
Grants and funding
- CTRNBC-2022/100001/CRUK_/Cancer Research UK/United Kingdom
- 17786/CRUK_/Cancer Research UK/United Kingdom
- 30025/CRUK_/Cancer Research UK/United Kingdom
- MR/L016311/1/MRC_/Medical Research Council/United Kingdom
- MR/P014712/1/MRC_/Medical Research Council/United Kingdom
- CC2041/WT_/Wellcome Trust/United Kingdom
- MR/V033077/1/MRC_/Medical Research Council/United Kingdom
- 29569/CRUK_/Cancer Research UK/United Kingdom
- MR/W025051/1/MRC_/Medical Research Council/United Kingdom
- 21999/CRUK_/Cancer Research UK/United Kingdom
- 24956/CRUK_/Cancer Research UK/United Kingdom
- CC2008/WT_/Wellcome Trust/United Kingdom
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
