

The evolution of lung cancer and impact of subclonal selection in TRACERx
- PMID: 37046096
- PMCID: PMC10115649
- DOI: 10.1038/s41586-023-05783-5
The evolution of lung cancer and impact of subclonal selection in TRACERx
Erratum in
-
Author Correction: The evolution of lung cancer and impact of subclonal selection in TRACERx.Nature. 2024 Jul;631(8022):E15. doi: 10.1038/s41586-024-07738-w. Nature. 2024. PMID: 38965439 Free PMC article. No abstract available.
Abstract
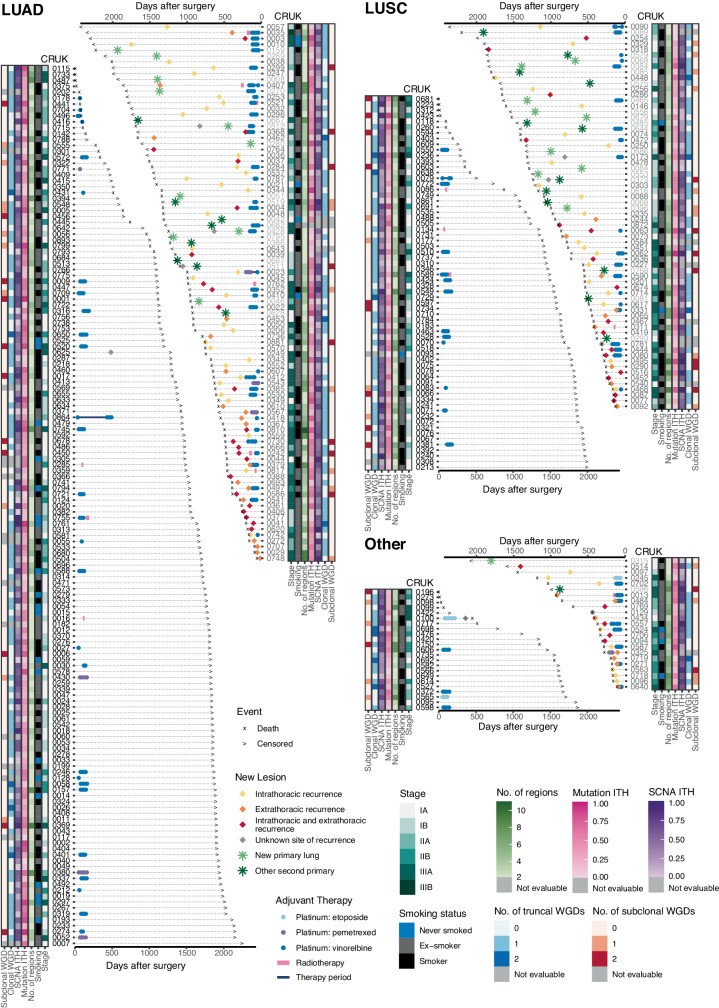
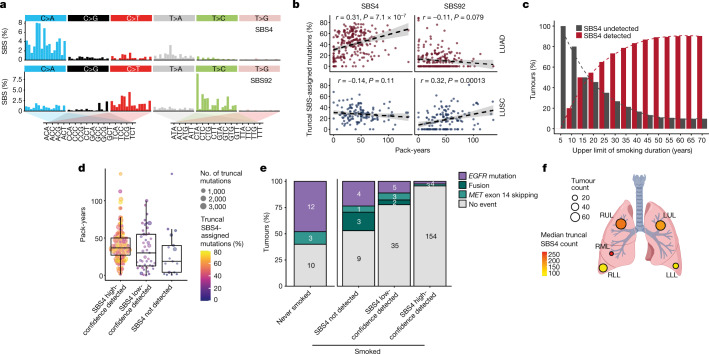
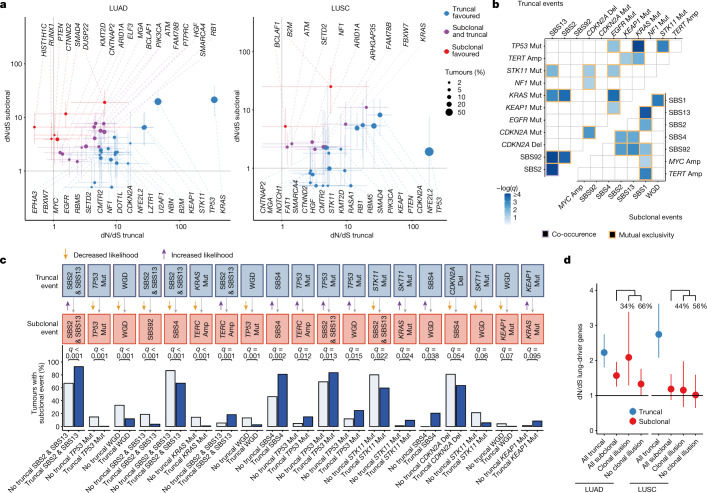
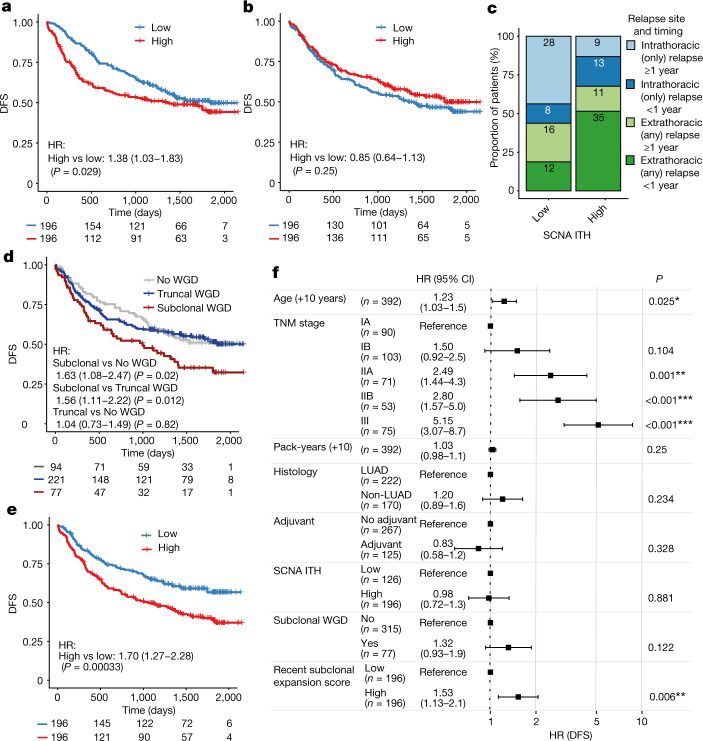
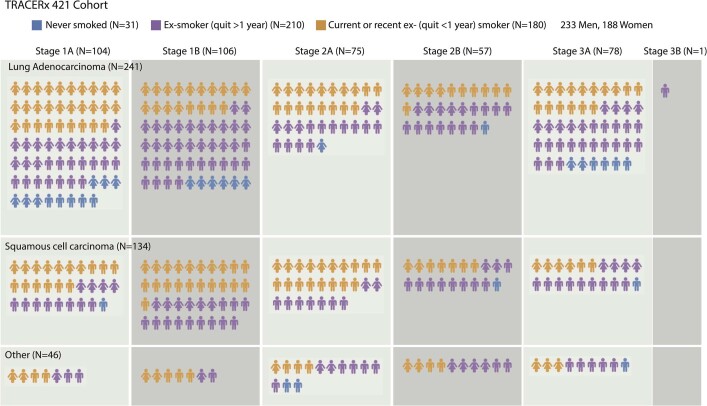
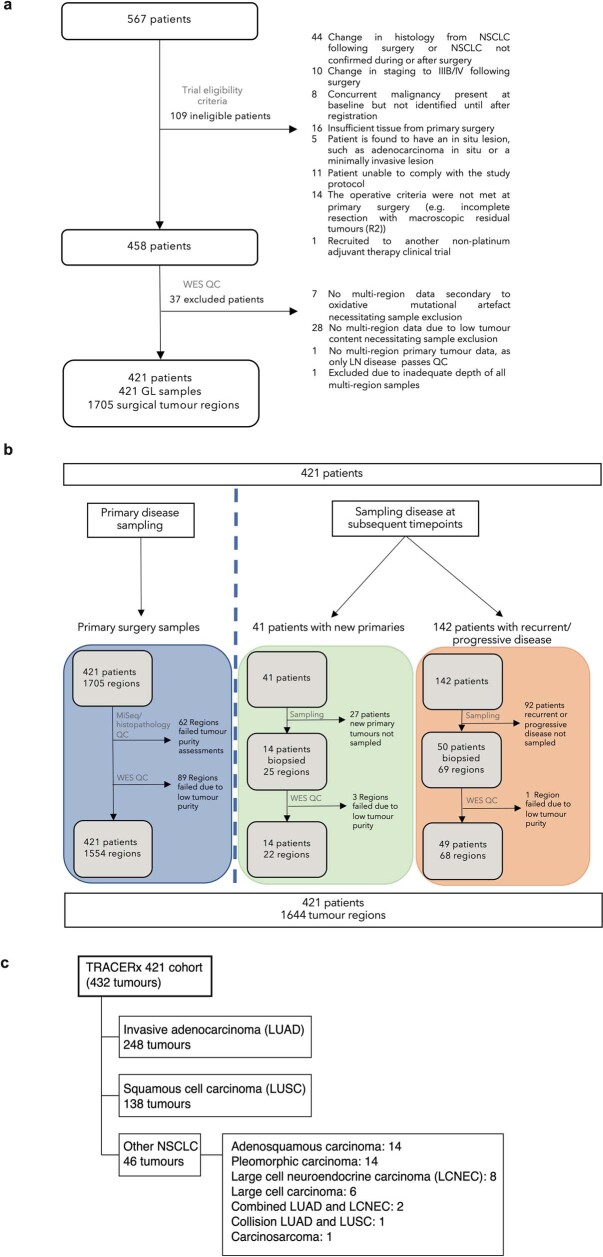
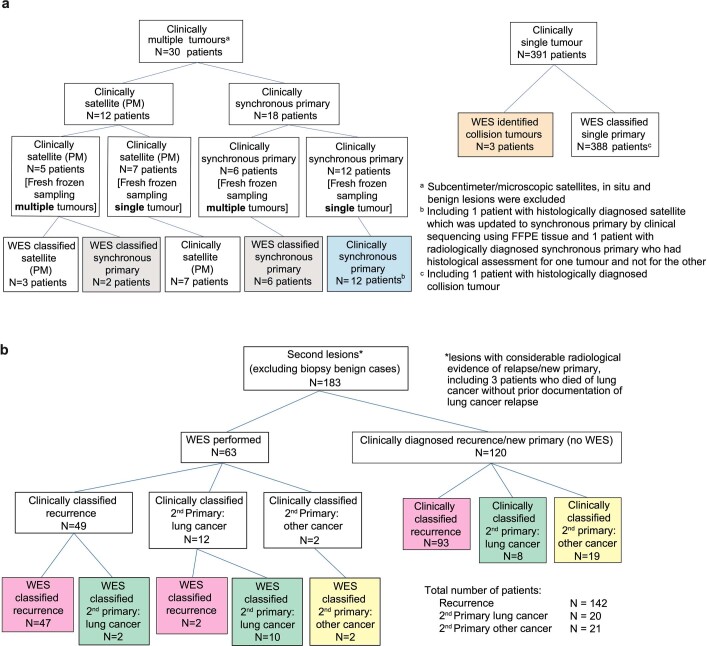
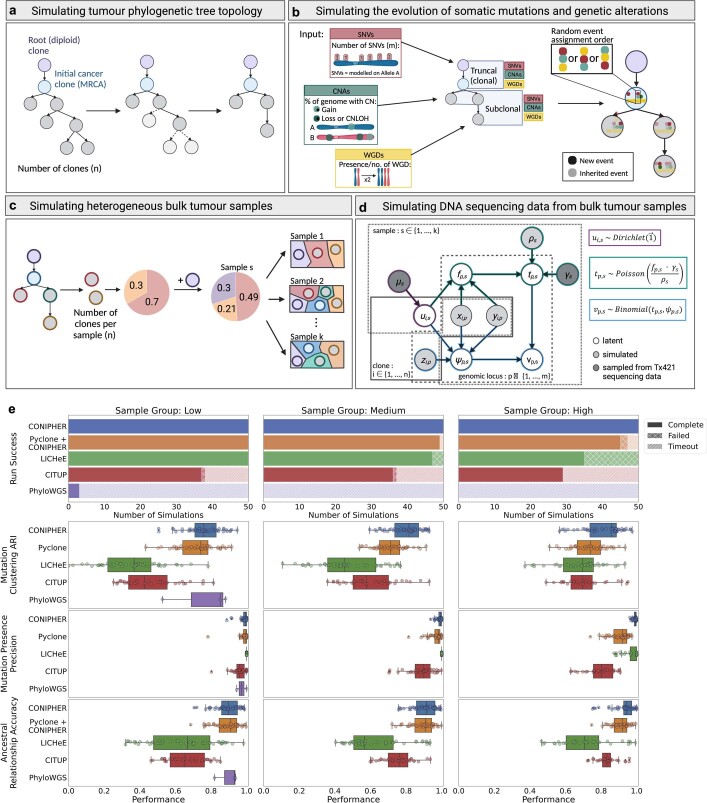
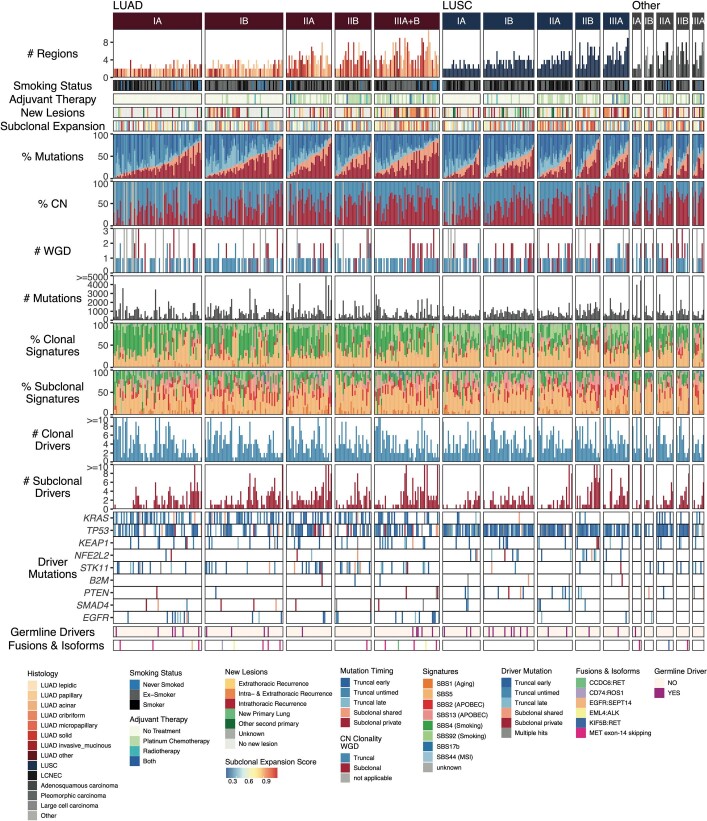
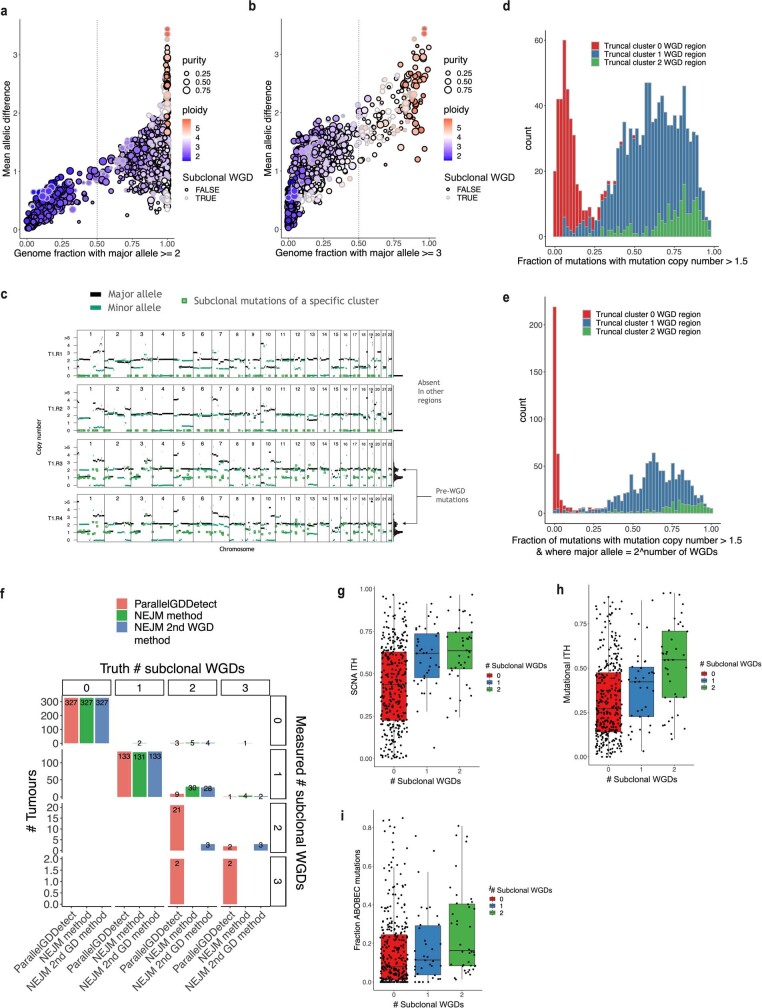
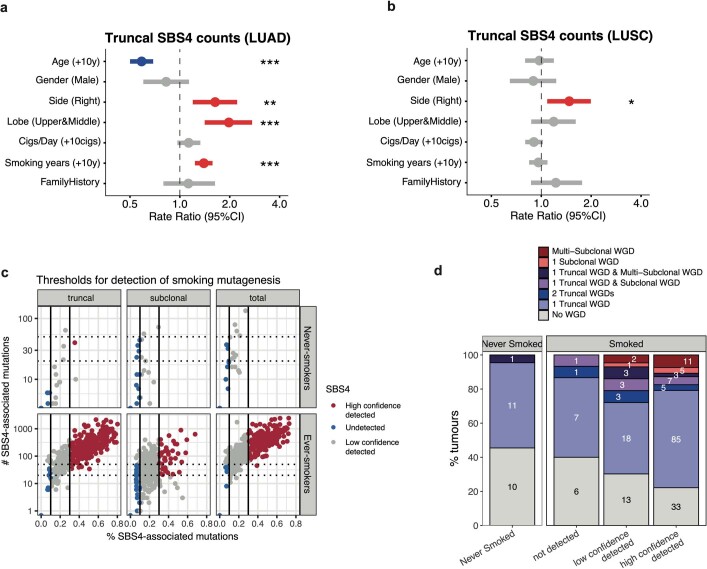
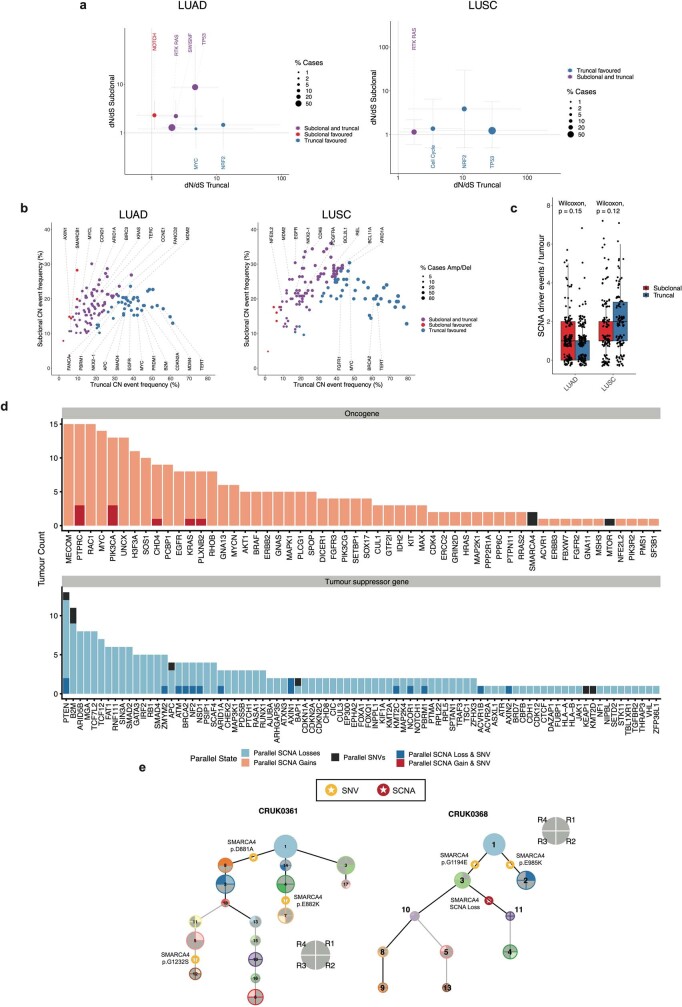
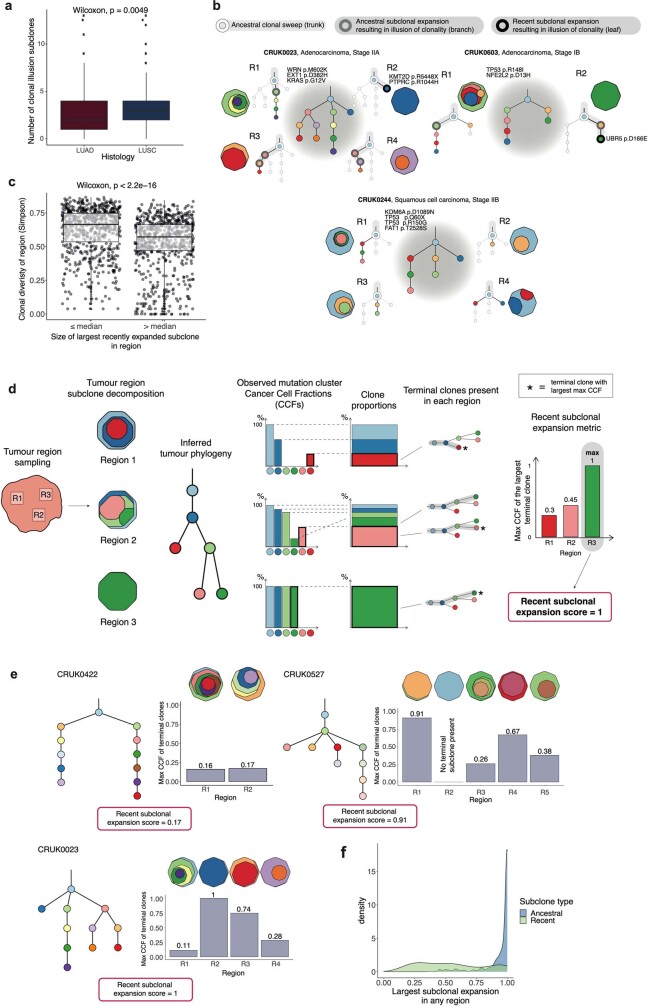
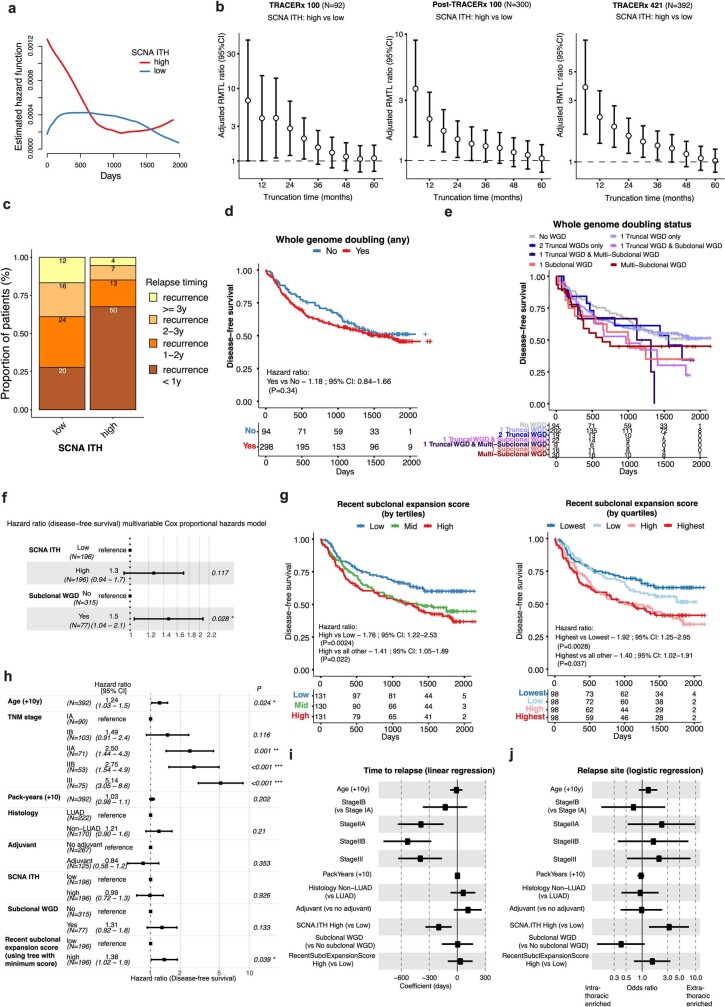
Lung cancer is the leading cause of cancer-associated mortality worldwide1. Here we analysed 1,644 tumour regions sampled at surgery or during follow-up from the first 421 patients with non-small cell lung cancer prospectively enrolled into the TRACERx study. This project aims to decipher lung cancer evolution and address the primary study endpoint: determining the relationship between intratumour heterogeneity and clinical outcome. In lung adenocarcinoma, mutations in 22 out of 40 common cancer genes were under significant subclonal selection, including classical tumour initiators such as TP53 and KRAS. We defined evolutionary dependencies between drivers, mutational processes and whole genome doubling (WGD) events. Despite patients having a history of smoking, 8% of lung adenocarcinomas lacked evidence of tobacco-induced mutagenesis. These tumours also had similar detection rates for EGFR mutations and for RET, ROS1, ALK and MET oncogenic isoforms compared with tumours in never-smokers, which suggests that they have a similar aetiology and pathogenesis. Large subclonal expansions were associated with positive subclonal selection. Patients with tumours harbouring recent subclonal expansions, on the terminus of a phylogenetic branch, had significantly shorter disease-free survival. Subclonal WGD was detected in 19% of tumours, and 10% of tumours harboured multiple subclonal WGDs in parallel. Subclonal, but not truncal, WGD was associated with shorter disease-free survival. Copy number heterogeneity was associated with extrathoracic relapse within 1 year after surgery. These data demonstrate the importance of clonal expansion, WGD and copy number instability in determining the timing and patterns of relapse in non-small cell lung cancer and provide a comprehensive clinical cancer evolutionary data resource.
Trial registration: ClinicalTrials.gov NCT01888601.
© 2023. The Author(s).
Conflict of interest statement
A.M.F. is a co-inventor on a patent application to determine methods and systems for tumour monitoring (PCT/EP2022/077987). M.A.B. has consulted for Achilles Therapeutics. D.A.M. reports speaker fees from AstraZeneca, Eli Lilly and Takeda, consultancy fees from AstraZeneca, Thermo Fisher, Takeda, Amgen, Janssen, MIM Software, Bristol-Myers Squibb (BMS) and Eli Lilly and has received educational support from Takeda and Amgen. S.V. is a co-inventor on a patent to detect molecules in a sample (US patent 10578620). G.A.W. is employed by and has stock options in Achilles Therapeutics. R.S. reports non-financial support from Merck and BMS, research support from Merck, Puma Biotechnology and Roche, and personal fees from Roche, BMS and Exact Sciences for advisory boards. C.A. has received speaking honoraria or expenses from Novartis, Roche, AstraZeneca and BMS and reports employment at AstraZeneca. C.A. is an inventor on a European patent application relating to assay technology to detect tumour recurrence (PCT/GB2017/053289); the patent has been licensed to commercial entities and under their terms of employment, C.A. is due a revenue share of any revenue generated from such licence(s). C.A. declares a patent application (PCT/US2017/028013) for methods to detect lung cancer. C.A. is a named inventor on a patent application to determine methods and systems for tumour monitoring (PCT/EP2022/077987). C.A. is a named inventor on a provisional patent protection related to a ctDNA detection algorithm. D.B. reports personal fees from NanoString and AstraZeneca. He has a patent (PCT/GB2020/050221) issued on methods for cancer prognostication. R.R. is an employee of and has stock options in Achilles Therapeutics and holds a European patent on targeting neoantigens (PCT/EP2016/059401) and in determining HLA LOH (PCT/GB2018/052004). D.A.F. reports grants from Aldeyra, Boehringer Ingelheim, Astex Therapeutics, Bayer, BMS, GSK, RS Oncology, Clovis, Eli Lilly, MSD, GSK, personal fees from Atara, BMS, Boehringer Ingelheim, Cambridge Clinical Laboratories, Targovax, Roche and RS Oncology. M.D.F. acknowledges grant support from CRUK, AstraZeneca, Boehringer Ingelheim, MSD and Merck; is an advisory board member for Transgene; and has consulted for Achilles, Amgen, AstraZeneca, Bayer, Boxer, BMS, Celgene, EQRx, Guardant Health, Immutep, Ixogen, Janssen, Merck, MSD, Nanobiotix, Novartis, Oxford VacMedix, Pharmamar, Pfizer, Roche, Takeda and UltraHuman. K.S.P. is a co-founder of Achilles Therapeutics. S.A.Q. is a co-founder, stockholder and Chief Scientific Officer of Achilles Therapeutics. E.L. has received funding from AstraZeneca, Boehringer Ingelheim, Medela, Johnson & Johnson/Ethicon, Covidien/Medtronic, Guardant Health, Takeda, Lilly Oncology, Boehringer Ingelheim and Bayer. E.L. has received consulting fees from Beigene, Roche and BMS, honoraria from Medela and is a founder My Cancer Companion, Healthcare Companion Ltd. N.N. reports honoraria for non-promotional educational talks, advisory boards or conference attendance from Amgen, AstraZeneca, Boehringer Ingelheim, BMS, Fujifilm, Guardant Health, Intuitive, Janssen, Lilly, Merck Sharp & Dohme, Olympus, OncLive, PeerVoice, Pfizer and Takeda. S.M.J. has received fees for advisory board membership in the past 3 years from AstraZeneca, Bard1 Lifescience and Johnson & Johnson. He has received a grant income from Owlstone and GRAIL Inc. He has received assistance with travel to an academic meeting from Cheisi. C.D. has received research funding and educational research grants from AstraZeneca, Astex Pharmaceuticals, Bioven, Amgen, Carrick Therapeutics, Merck, Taiho Oncology, GSK, Bayer, Boehringer Ingelheim, Roche, BMS, Novartis, Celgene, Epigene Therapeutics, Angle PLC, Menarini, Clearbridge Biomedics, Thermo Fisher Scientific and Neomed Therapeutics. C.D. has also received honoraria for consultancy and/or advisory boards from Biocartis, Merck, AstraZeneca, GRAIL and Boehringer Ingelheim. J.C. reports funding from Amgen to attend a conference. A. Hackshaw has received fees for being a member of Independent Data Monitoring Committees for Roche-sponsored clinical trials, and academic projects co-ordinated by Roche. N.J.B. is a co-inventor on a patent to identify responders to cancer treatment (PCT/GB2018/051912), has a patent application (PCT/GB2020/050221) on methods for cancer prognostication and is a co-inventor on a patent for methods for predicting anticancer responses (US14/466,208). C.T.H. has received speaker fees from AstraZeneca. M.J.-H. has consulted for and is a member of the Achilles Therapeutics Scientific Advisory Board (SAB) and Steering Committee, has received speaker honoraria from Pfizer, Astex Pharmaceuticals, Oslo Cancer Cluster, and holds a patent (PCT/US2017/028013) relating to methods for lung cancer detection. This patent has been licensed to commercial entities and under terms of employment, M.J.-H. is due a share of any revenue generated from such licence(s). N.M. has received consultancy fees and has stock options in Achilles Therapeutics. N.M. holds European patents relating to targeting neoantigens (PCT/EP2016/059401), identifying patient response to immune checkpoint blockade (PCT/EP2016/071471), determining HLA LOH (PCT/GB2018/052004) and predicting survival rates of patients with cancer (PCT/GB2020/050221). C.S. acknowledges grant support from AstraZeneca, Boehringer-Ingelheim, BMS, Pfizer, Roche-Ventana, Invitae (previously Archer Dx (collaboration in minimal residual disease sequencing technologies)) and Ono Pharmaceutical. C.S. is an AstraZeneca Advisory Board member and Chief Investigator for the AZ MeRmaiD 1 and 2 clinical trials and is also Co-Chief Investigator of the NHS Galleri trial funded by GRAIL and a paid member of GRAIL’s SAB. He receives consultant fees from Achilles Therapeutics (also a SAB member), Bicycle Therapeutics (also a SAB member), Genentech, Medicxi, Roche Innovation Centre–Shanghai, Metabomed (until July 2022), and the Sarah Cannon Research Institute. C.S. had stock options in Apogen Biotechnologies and GRAIL until June 2021, and currently has stock options in Epic Bioscience, Bicycle Therapeutics, and has stock options and is co-founder of Achilles Therapeutics. C.S. is an inventor on a European patent application relating to an assay technology to detect tumour recurrence (PCT/GB2017/053289), the patent has been licensed to commercial entities and under his terms of employment, C.S. is due a revenue share of any revenue generated from such licence(s). C.S. holds patents relating to targeting neoantigens (PCT/EP2016/059401), identifying patient responses to immune checkpoint blockade (PCT/EP2016/071471), determining HLA LOH (PCT/GB2018/052004), predicting survival rates of patients with cancer (PCT/GB2020/050221), identifying patients who respond to cancer treatment (PCT/GB2018/051912), a US patent relating to detecting tumour mutations (PCT/US2017/28013), methods for lung cancer detection (US20190106751A1) and both a European and US patent related to identifying indel mutation targets (PCT/GB2018/051892) and is a co-inventor on a patent application to determine methods and systems for tumour monitoring (PCT/EP2022/077987). C.S. is a named inventor on a provisional patent related to a ctDNA detection algorithm.
Figures
Comment in
-
Molecular portraits of lung cancer evolution.Nature. 2023 Apr;616(7957):435-436. doi: 10.1038/d41586-023-00934-0. Nature. 2023. PMID: 37045956 No abstract available.
References
Publication types
MeSH terms
Substances
Associated data
Grants and funding
- CTRNBC-2022/100001/CRUK_/Cancer Research UK/United Kingdom
- 17786/CRUK_/Cancer Research UK/United Kingdom
- 30025/CRUK_/Cancer Research UK/United Kingdom
- CC2041/WT_/Wellcome Trust/United Kingdom
- MR/V033077/1/MRC_/Medical Research Council/United Kingdom
- MR/T02481X/1/MRC_/Medical Research Council/United Kingdom
- 29569/CRUK_/Cancer Research UK/United Kingdom
- MR/W025051/1/MRC_/Medical Research Council/United Kingdom
- 21999/CRUK_/Cancer Research UK/United Kingdom
- 24956/CRUK_/Cancer Research UK/United Kingdom
- CC2008/WT_/Wellcome Trust/United Kingdom
- CTUQQR-DEC22/100009/CRUK_/Cancer Research UK/United Kingdom
- 25354/CRUK_/Cancer Research UK/United Kingdom
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
