

Signatures of Co-Deregulated Genes and Their Transcriptional Regulators in Kidney Cancers
- PMID: 37047552
- PMCID: PMC10094846
- DOI: 10.3390/ijms24076577
Signatures of Co-Deregulated Genes and Their Transcriptional Regulators in Kidney Cancers
Abstract
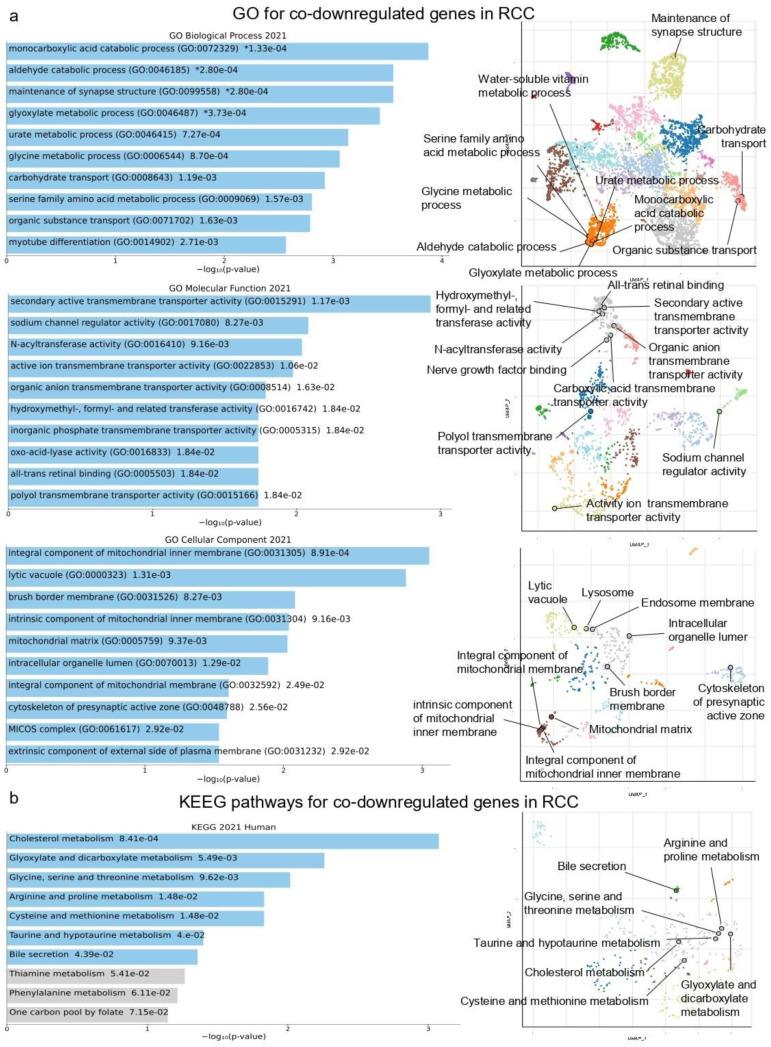
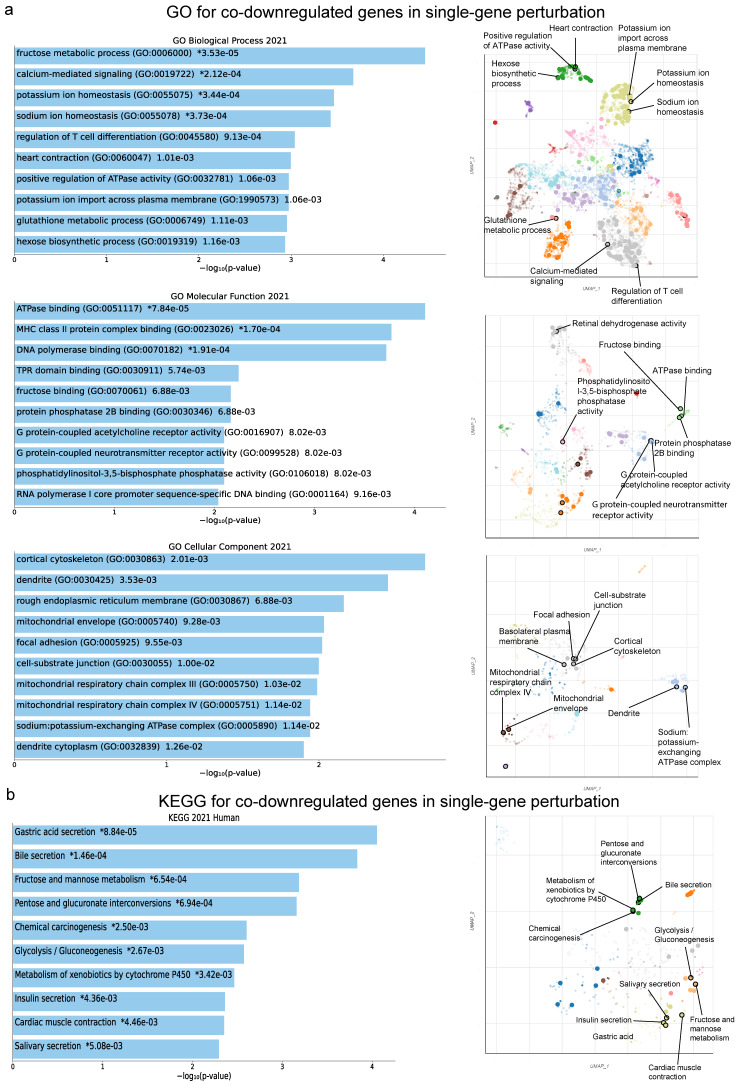
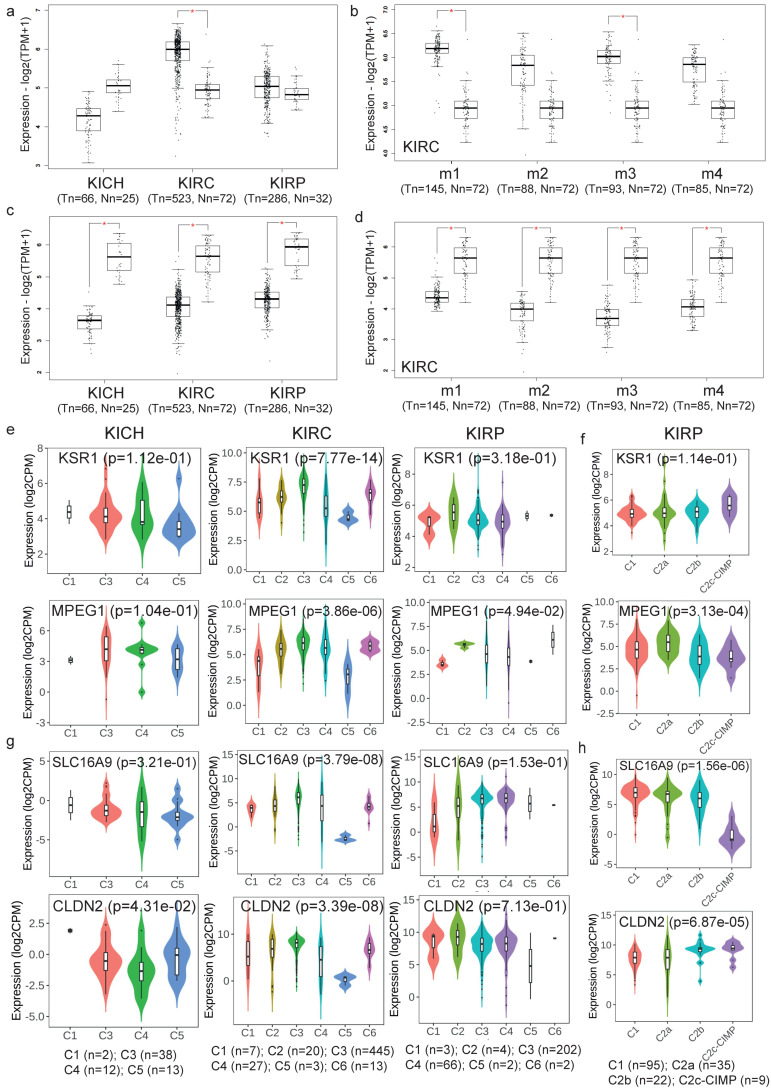
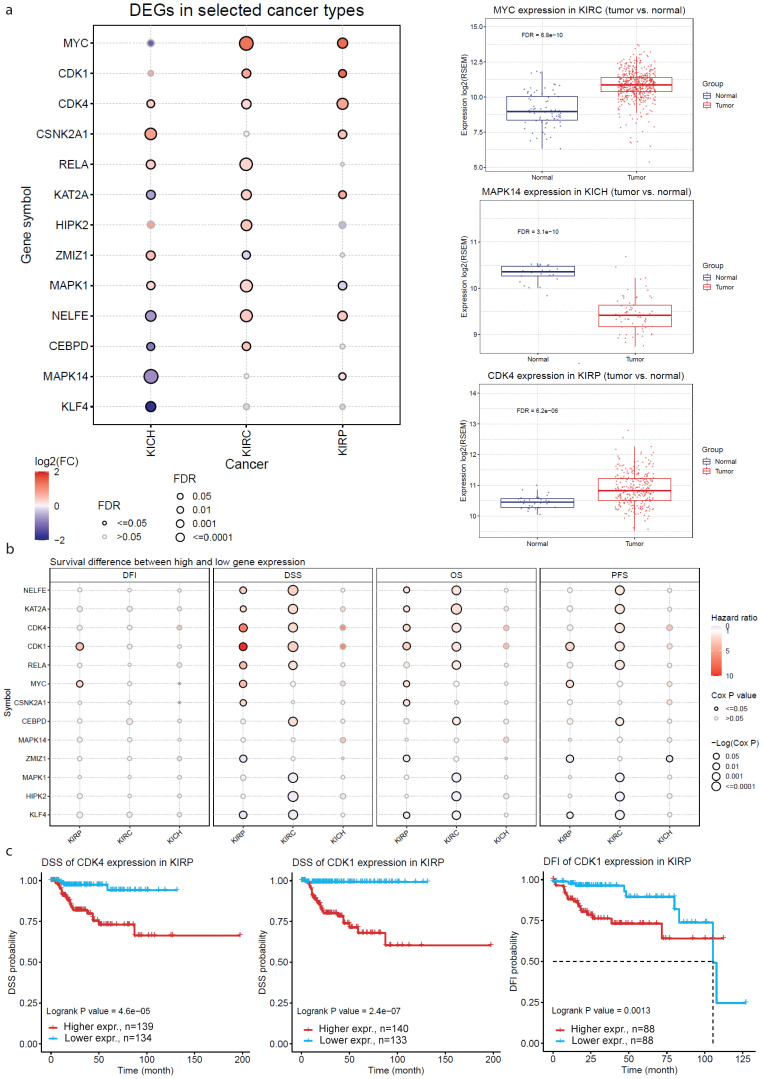
There are several studies on the deregulated gene expression profiles in kidney cancer, with varying results depending on the tumor histology and other parameters. None of these, however, have identified the networks that the co-deregulated genes (co-DEGs), across different studies, create. Here, we reanalyzed 10 Gene Expression Omnibus (GEO) studies to detect and annotate co-deregulated signatures across different subtypes of kidney cancer or in single-gene perturbation experiments in kidney cancer cells and/or tissue. Using a systems biology approach, we aimed to decipher the networks they form along with their upstream regulators. Differential expression and upstream regulators, including transcription factors [MYC proto-oncogene (MYC), CCAAT enhancer binding protein delta (CEBPD), RELA proto-oncogene, NF-kB subunit (RELA), zinc finger MIZ-type containing 1 (ZMIZ1), negative elongation factor complex member E (NELFE) and Kruppel-like factor 4 (KLF4)] and protein kinases [Casein kinase 2 alpha 1 (CSNK2A1), mitogen-activated protein kinases 1 (MAPK1) and 14 (MAPK14), Sirtuin 1 (SIRT1), Cyclin dependent kinases 1 (CDK1) and 4 (CDK4), Homeodomain interacting protein kinase 2 (HIPK2) and Extracellular signal-regulated kinases 1 and 2 (ERK1/2)], were computed using the Characteristic Direction, as well as GEO2Enrichr and X2K, respectively, and further subjected to GO and KEGG pathways enrichment analyses. Furthermore, using CMap, DrugMatrix and the LINCS L1000 chemical perturbation databases, we highlight putative repurposing drugs, including Etoposide, Haloperidol, BW-B70C, Triamterene, Chlorphenesin, BRD-K79459005 and β-Estradiol 3-benzoate, among others, that may reverse the expression of the identified co-DEGs in kidney cancers. Of these, the cytotoxic effects of Etoposide, Catecholamine, Cyclosporin A, BW-B70C and Lasalocid sodium were validated in vitro. Overall, we identified critical co-DEGs across different subtypes in kidney cancer, and our results provide an innovative framework for their potential use in the future.
Keywords: Characteristic Direction; GEO2Enrichr; KICH; KIRC; KIRP; X2K; co-deregulated genes; drug repurposing; kidney cancer; single-drug perturbation; single-gene perturbation; tumor heterogeneity.
Conflict of interest statement
The authors declare no conflict of interest.
Figures








Similar articles
-
Signatures of Co-Deregulated Genes and Their Transcriptional Regulators in Lung Cancer.Int J Mol Sci. 2022 Sep 18;23(18):10933. doi: 10.3390/ijms231810933. Int J Mol Sci. 2022. PMID: 36142846 Free PMC article.
-
Signatures of co-deregulated genes and their transcriptional regulators in colorectal cancer.NPJ Syst Biol Appl. 2020 Jul 31;6(1):23. doi: 10.1038/s41540-020-00144-8. NPJ Syst Biol Appl. 2020. PMID: 32737302 Free PMC article.
-
System analysis of VEGFA in renal cell carcinoma: The expression, prognosis, gene regulation network and regulation targets.Int J Biol Markers. 2022 Mar;37(1):90-101. doi: 10.1177/17246008211063501. Epub 2021 Dec 6. Int J Biol Markers. 2022. PMID: 34870494
-
Good or not good: Role of miR-18a in cancer biology.Rep Pract Oncol Radiother. 2020 Sep-Oct;25(5):808-819. doi: 10.1016/j.rpor.2020.07.006. Epub 2020 Aug 12. Rep Pract Oncol Radiother. 2020. PMID: 32884453 Free PMC article. Review.
-
Gene methylation in gastric cancer.Clin Chim Acta. 2013 Sep 23;424:53-65. doi: 10.1016/j.cca.2013.05.002. Epub 2013 May 10. Clin Chim Acta. 2013. PMID: 23669186 Review.
Cited by
-
CENPT prevents renal cell carcinoma against ferroptosis by enhancing the synthesis of glutathione.Cell Death Dis. 2025 Jul 12;16(1):517. doi: 10.1038/s41419-025-07848-x. Cell Death Dis. 2025. PMID: 40651948 Free PMC article.
-
Single-cell disulfidptosis regulator patterns guide intercellular communication of tumor microenvironment that contribute to kidney renal clear cell carcinoma progression and immunotherapy.Front Immunol. 2024 Jan 16;15:1288240. doi: 10.3389/fimmu.2024.1288240. eCollection 2024. Front Immunol. 2024. PMID: 38292868 Free PMC article.
-
Exploration of the Mechanisms Underlying Yu's Enema Formula in Treating Ulcerative Colitis by Blocking the RhoA/ROCK Pathway based on Network Pharmacology, High-performance Liquid Chromatography Analysis, and Experimental Verification.Curr Pharm Des. 2024;30(14):1085-1102. doi: 10.2174/0113816128290586240315071044. Curr Pharm Des. 2024. PMID: 38523541
References
-
- Safiri S., Kolahi A.-A., Mansournia M.A., Almasi-Hashiani A., Ashrafi-Asgarabad A., Sullman M.J.M., Bettampadi D., Qorbani M., Moradi-Lakeh M., Ardalan M., et al. The Burden of Kidney Cancer and Its Attributable Risk Factors in 195 Countries and Territories, 1990–2017. Sci. Rep. 2020;10:13862. doi: 10.1038/s41598-020-70840-2. - DOI - PMC - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
