

New Insight into the Genotype-Phenotype Correlation of PRPH2-Related Diseases Based on a Large Chinese Cohort and Literature Review
- PMID: 37047703
- PMCID: PMC10095211
- DOI: 10.3390/ijms24076728
New Insight into the Genotype-Phenotype Correlation of PRPH2-Related Diseases Based on a Large Chinese Cohort and Literature Review
Abstract
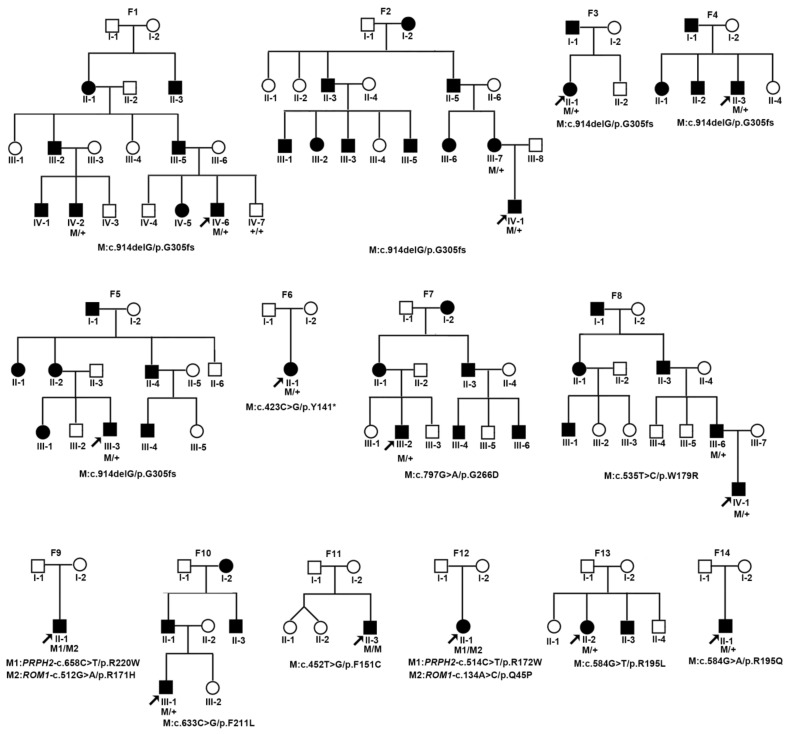
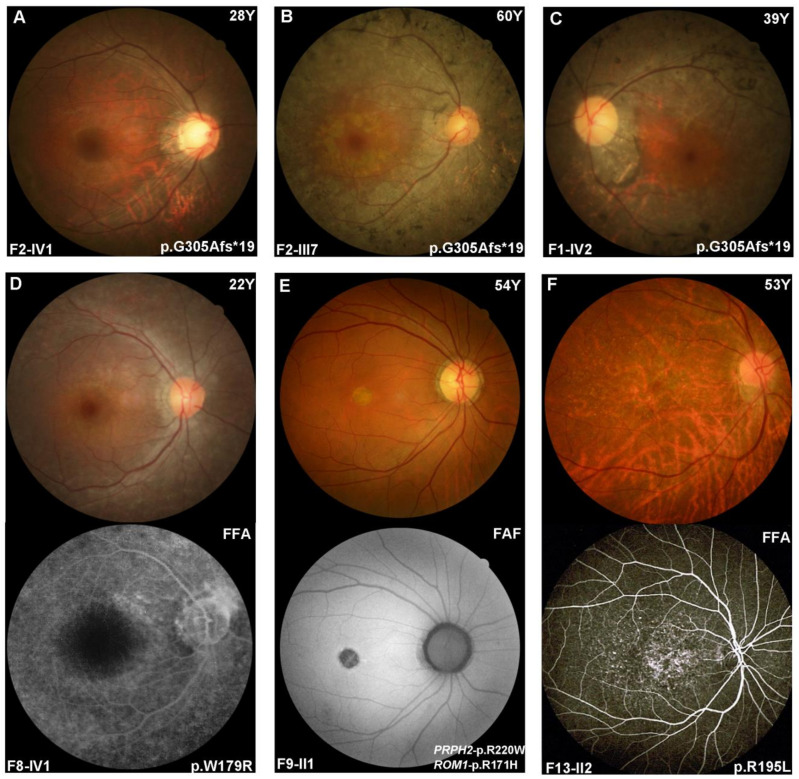
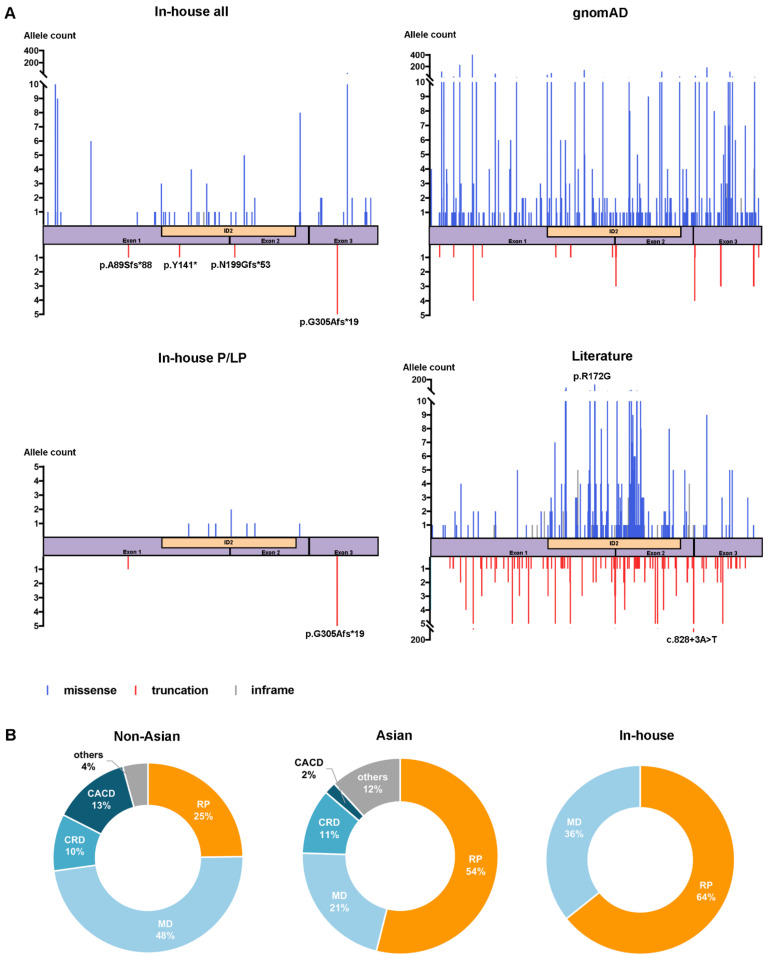
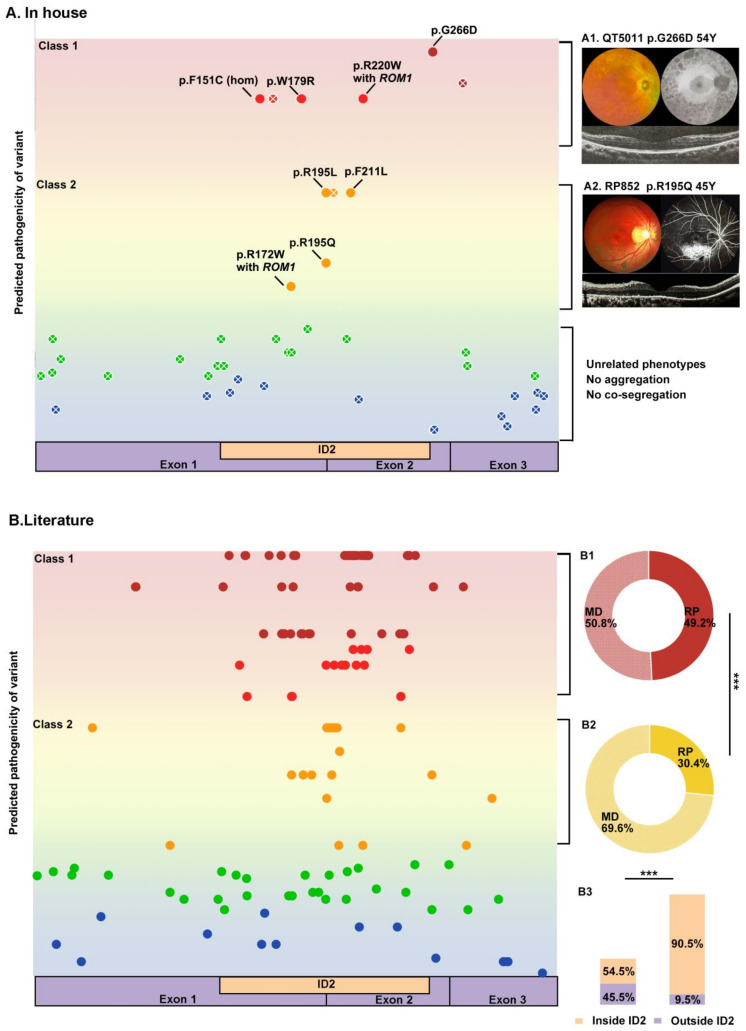
Variants in PRPH2 are a common cause of inherited retinal dystrophies with high genetic and phenotypic heterogeneity. In this study, variants in PRPH2 were selected from in-house exome sequencing data, and all reported PRPH2 variants were evaluated with the assistance of online prediction tools and the comparative validation of large datasets. All variants were classified based on the American College of Medical Genetics and Genomics/Association for Molecular Pathology (ACMG/AMP) guidelines. Individuals with pathogenic or likely pathogenic variants of PRPH2 were confirmed by Sanger sequencing. Clinical characteristics were summarized. Ten pathogenic or likely pathogenic variants of PRPH2 were identified in 14 families. In our cohort, the most frequent variant was p.G305Afs*19, accounting for 33.3% (5/15) of alleles, in contrast to the literature, where p.R172G (11.6%, 119/1028) was the most common variant. Nine in-house families (63.8%) were diagnosed with retinitis pigmentosa (RP), distinct from the phenotypic spectrum in the literature, which shows that RP accounts for 27.9% (283/1013) and macular degeneration is more common (45.2%, 458/1013). Patients carrying missense variants predicted as damaging by all seven prediction tools and absent in the gnomAD database were more likely to develop RP compared to those carrying missense variants predicted as damaging with fewer tools or with more than one allele number in the gnomAD database (p = 0.001). The population-specific genetic and phenotypic spectra of PRPH2 were explored, and novel insight into the genotype-phenotype correlation of PRPH2 was proposed. These findings demonstrated the importance of assessing PRPH2 variants in distinct populations and the value of providing practical suggestions for the genetic interpretation of PRPH2 variants.
Keywords: PRPH2; genotype-phenotype correlation; macular degeneration; retinitis pigmentosa.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
PRPH2-Related Retinal Dystrophies: Mutational Spectrum in 103 Families from a Spanish Cohort.Int J Mol Sci. 2024 Mar 2;25(5):2913. doi: 10.3390/ijms25052913. Int J Mol Sci. 2024. PMID: 38474159 Free PMC article.
-
Unveiling the Genetic and Phenotypic Landscape of a Chinese Cohort With Retinitis Pigmentosa.Mol Genet Genomic Med. 2025 Feb;13(2):e70011. doi: 10.1002/mgg3.70011. Mol Genet Genomic Med. 2025. PMID: 39988772 Free PMC article.
-
Autosomal dominant retinitis pigmentosa with macular involvement associated with a disease haplotype that included a novel PRPH2 variant (p.Cys250Gly).Ophthalmic Genet. 2018 Jun;39(3):357-365. doi: 10.1080/13816810.2018.1459737. Epub 2018 Apr 9. Ophthalmic Genet. 2018. PMID: 29630435
-
Characterizing the genotypic spectrum of retinitis pigmentosa in East Asian populations: a systematic review.Ophthalmic Genet. 2023 Apr;44(2):109-118. doi: 10.1080/13816810.2023.2182329. Epub 2023 Mar 1. Ophthalmic Genet. 2023. PMID: 36856324
-
Gene therapy for PRPH2-associated ocular disease: challenges and prospects.Cold Spring Harb Perspect Med. 2014 Aug 28;4(11):a017376. doi: 10.1101/cshperspect.a017376. Cold Spring Harb Perspect Med. 2014. PMID: 25167981 Free PMC article. Review.
Cited by
-
Expanding Genetic and Clinical Spectra of Inherited Retinal Dystrophies: Identification of Three Novel PRPH2 Variants.Biomedicines. 2025 Jun 23;13(7):1531. doi: 10.3390/biomedicines13071531. Biomedicines. 2025. PMID: 40722607 Free PMC article.
-
Current understanding on Retinitis Pigmentosa: a literature review.Front Ophthalmol (Lausanne). 2025 Jun 12;5:1600283. doi: 10.3389/fopht.2025.1600283. eCollection 2025. Front Ophthalmol (Lausanne). 2025. PMID: 40575766 Free PMC article. Review.
-
PRPH2-Related Retinal Dystrophies: Mutational Spectrum in 103 Families from a Spanish Cohort.Int J Mol Sci. 2024 Mar 2;25(5):2913. doi: 10.3390/ijms25052913. Int J Mol Sci. 2024. PMID: 38474159 Free PMC article.
-
ABCA4 Deep Intronic Variants Contributed to Nearly Half of Unsolved Stargardt Cases With a Milder Phenotype.Invest Ophthalmol Vis Sci. 2025 Jan 2;66(1):65. doi: 10.1167/iovs.66.1.65. Invest Ophthalmol Vis Sci. 2025. PMID: 39883546 Free PMC article.
-
Phenotypic and Genetic Heterogeneity of a Pakistani Cohort of 15 Consanguineous Families Segregating Variants in Leber Congenital Amaurosis-Associated Genes.Genes (Basel). 2024 Dec 21;15(12):1646. doi: 10.3390/genes15121646. Genes (Basel). 2024. PMID: 39766915 Free PMC article.
References
-
- Molday R.S., Hicks D., Molday L. Peripherin. A rim-specific membrane protein of rod outer segment discs. Investig. Ophthalmol. Vis. Sci. 1987;28:50–61. - PubMed
-
- Pontikos N., Arno G., Jurkute N., Schiff E., Ba-Abbad R., Malka S., Gimenez A., Georgiou M., Wright G., Armengol M., et al. Genetic Basis of Inherited Retinal Disease in a Molecularly Characterized Cohort of More Than 3000 Families from the United Kingdom. Ophthalmology. 2020;127:1384–1394. doi: 10.1016/j.ophtha.2020.04.008. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
