

Microglia Mediated Neuroinflammation in Parkinson's Disease
- PMID: 37048085
- PMCID: PMC10093562
- DOI: 10.3390/cells12071012
Microglia Mediated Neuroinflammation in Parkinson's Disease
Abstract
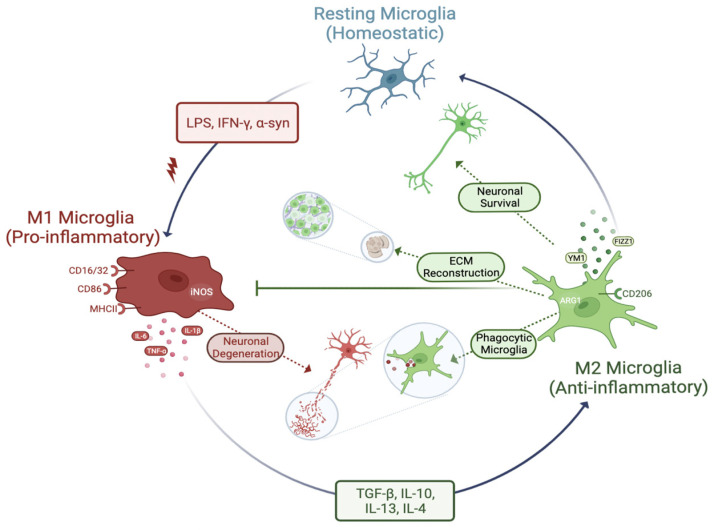
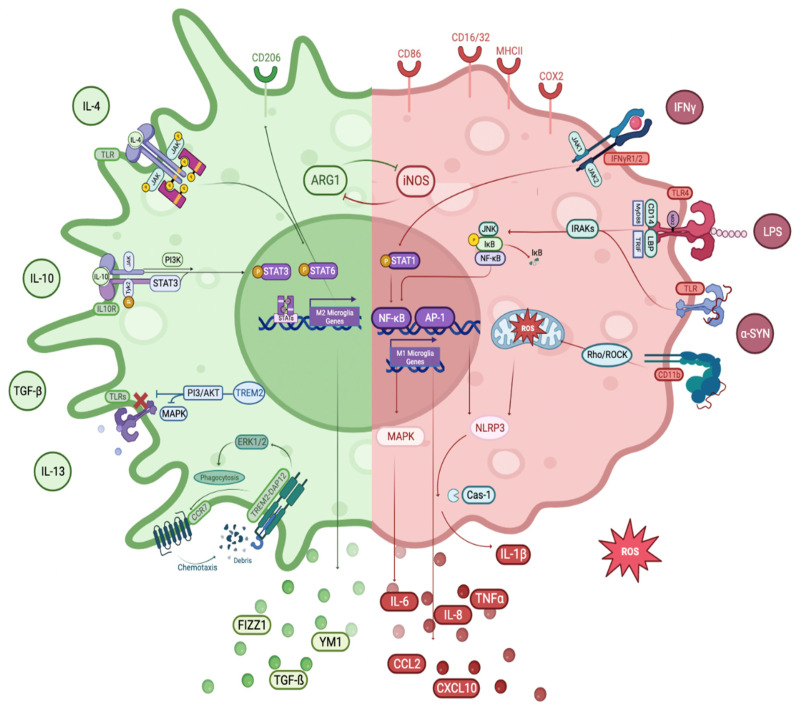
Parkinson's Disease (PD) is the second most common neurodegenerative disorder seen, especially in the elderly. Tremor, shaking, movement problems, and difficulty with balance and coordination are among the hallmarks, and dopaminergic neuronal loss in substantia nigra pars compacta of the brain and aggregation of intracellular protein α-synuclein are the pathological characterizations. Neuroinflammation has emerged as an involving mechanism at the initiation and development of PD. It is a complex network of interactions comprising immune and non-immune cells in addition to mediators of the immune response. Microglia, the resident macrophages in the CNS, take on the leading role in regulating neuroinflammation and maintaining homeostasis. Under normal physiological conditions, they exist as "homeostatic" but upon pathological stimuli, they switch to the "reactive state". Pro-inflammatory (M1) and anti-inflammatory (M2) phenotypes are used to classify microglial activity with each phenotype having its own markers and released mediators. When M1 microglia are persistent, they will contribute to various inflammatory diseases, including neurodegenerative diseases, such as PD. In this review, we focus on the role of microglia mediated neuroinflammation in PD and also signaling pathways, receptors, and mediators involved in the process, presenting the studies that associate microglia-mediated inflammation with PD. A better understanding of this complex network and interactions is important in seeking new therapies for PD and possibly other neurodegenerative diseases.
Keywords: M1 phenotype; M2 phenotype; Parkinson’s Disease; anti-inflammatory phenotype; microglial activation; neuroinflammation; α-synuclein.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Frost J.J., Rosier A.J., Reich S.G., Smith J.S., Ehlers M.D., Snyder S.H., Ravert H.T., Dannals R.F. Positron Emission Tomographic Imaging of the Dopamine Transporter with 11C-WIN 35,428 Reveals Marked Declines in Mild Parkinson’s Disease. Ann. Neurol. 1993;34:423–431. doi: 10.1002/ana.410340331. - DOI - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
