

High Glucose Activates Prolyl Hydroxylases and Disrupts HIF-α Signaling via the P53/TIGAR Pathway in Cardiomyocyte
- PMID: 37048134
- PMCID: PMC10093703
- DOI: 10.3390/cells12071060
High Glucose Activates Prolyl Hydroxylases and Disrupts HIF-α Signaling via the P53/TIGAR Pathway in Cardiomyocyte
Abstract
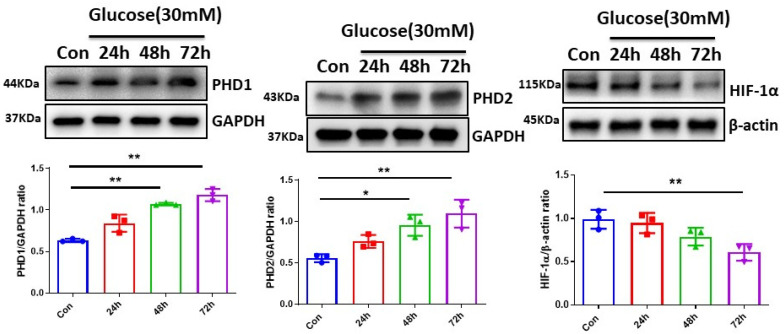
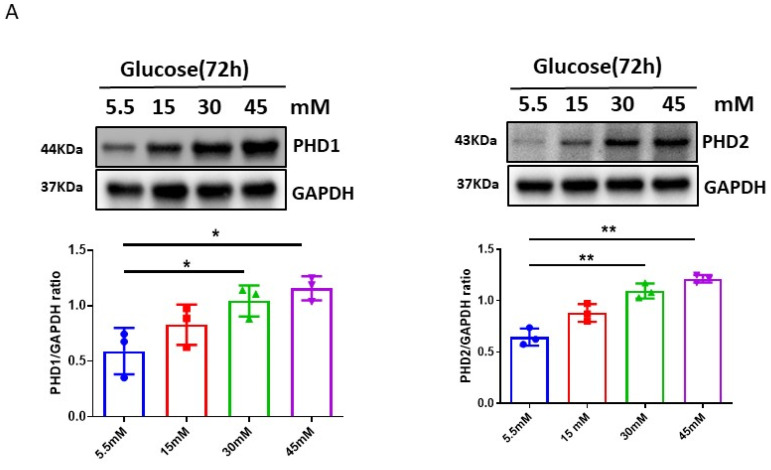
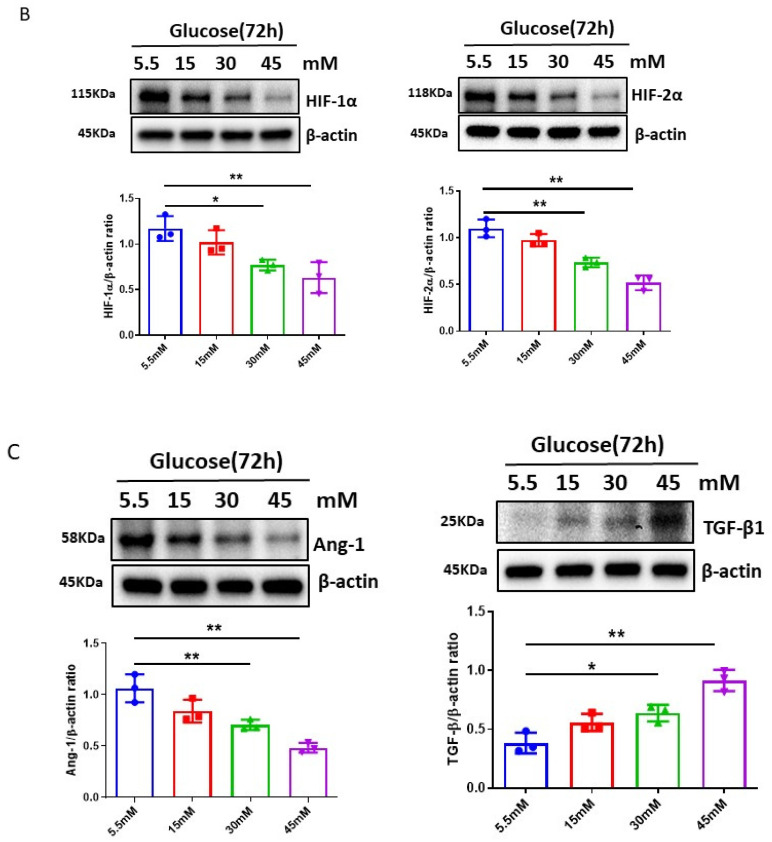
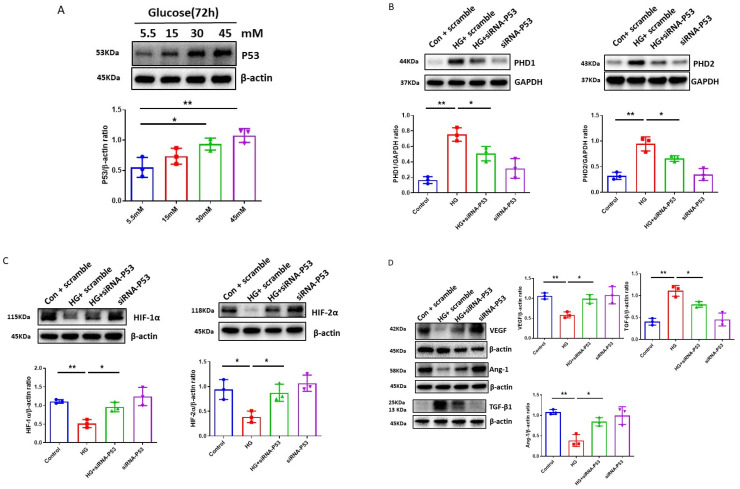
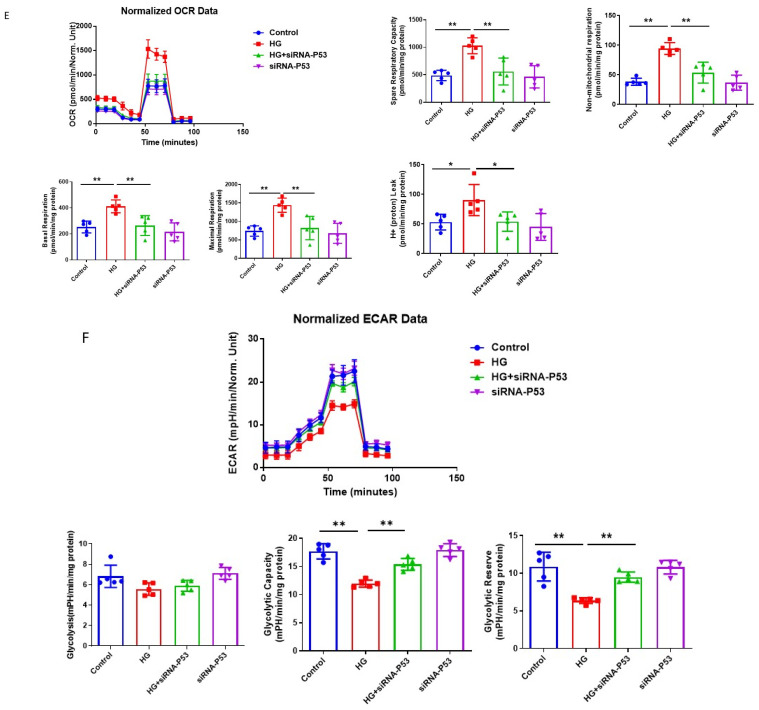
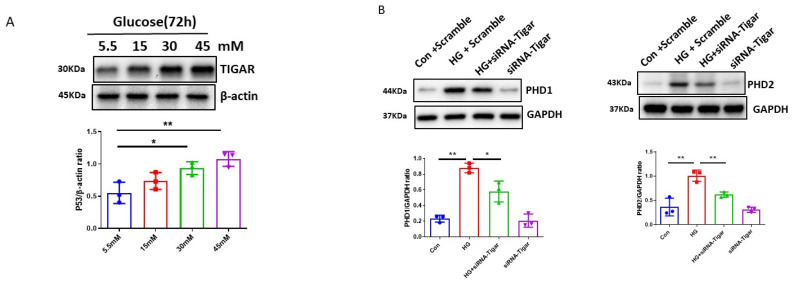
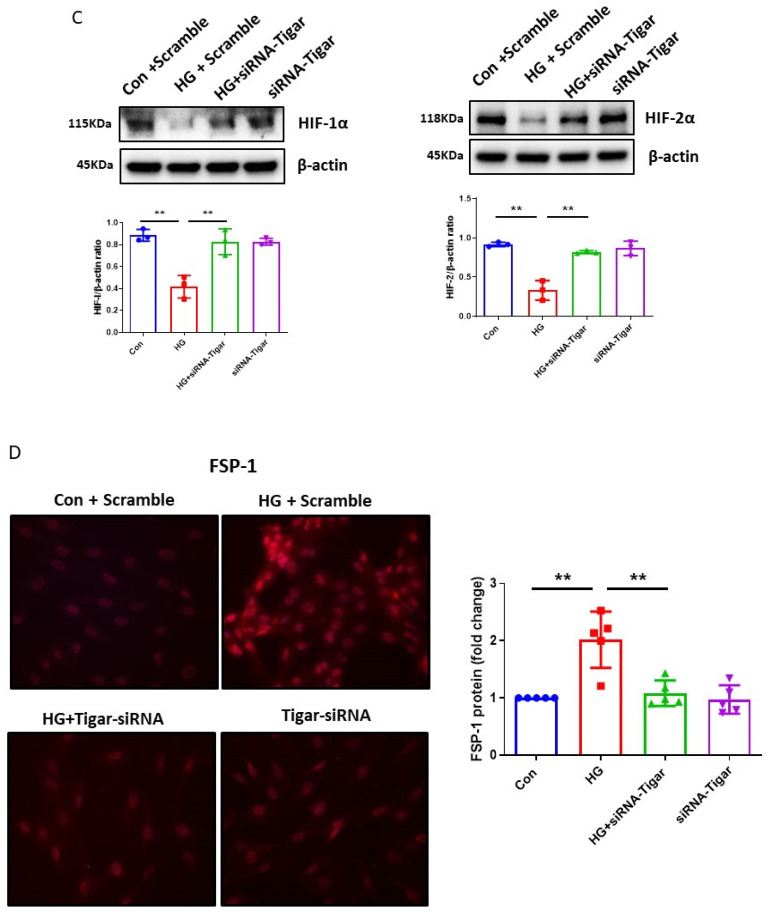
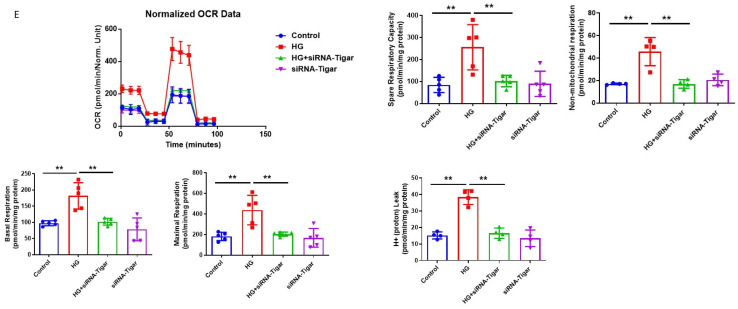
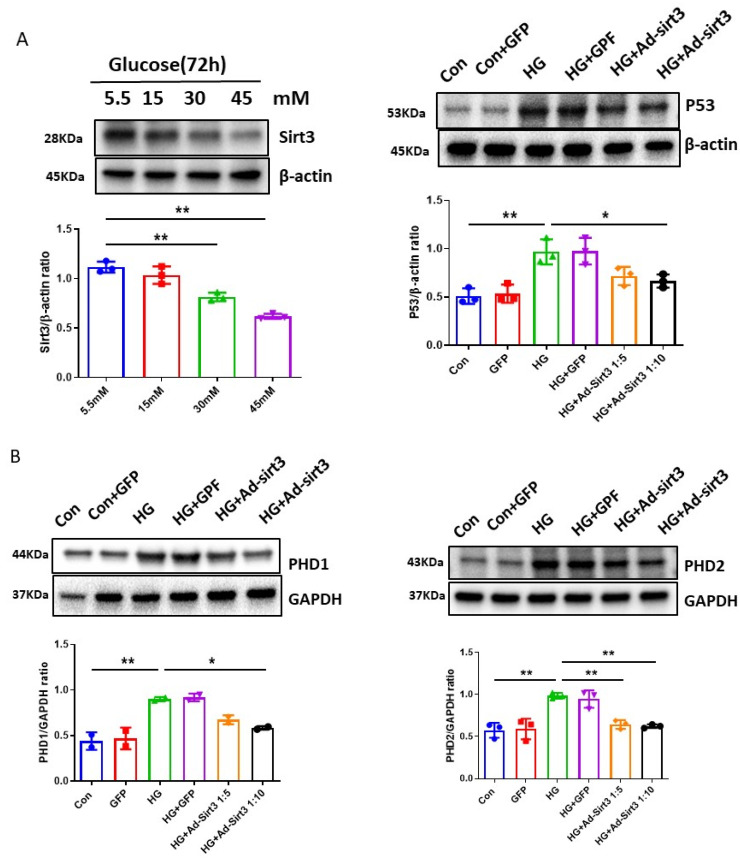
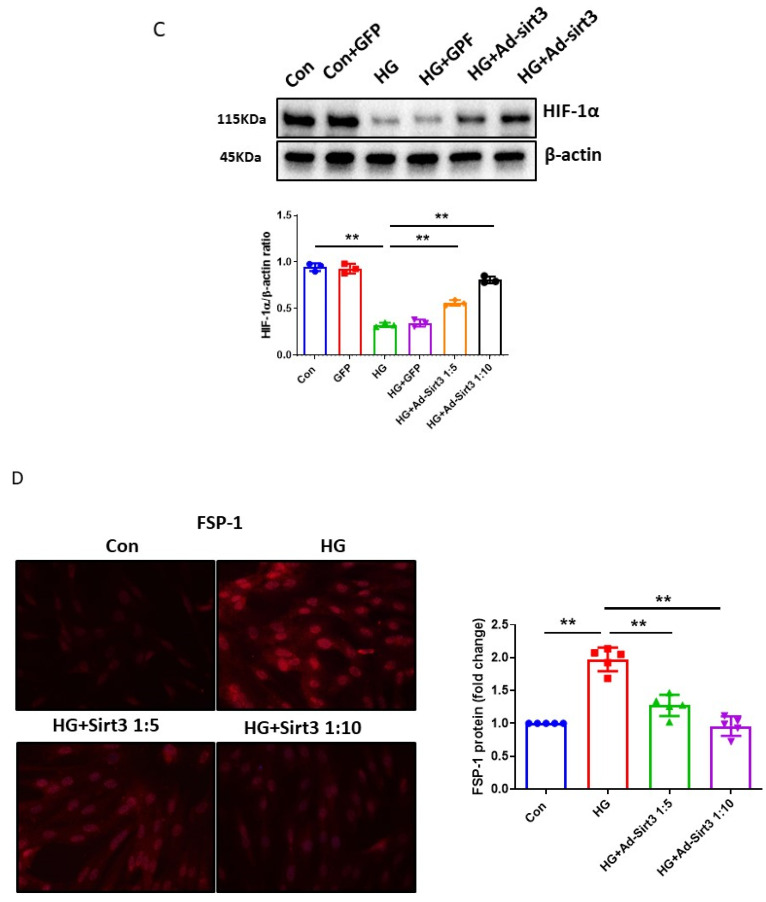
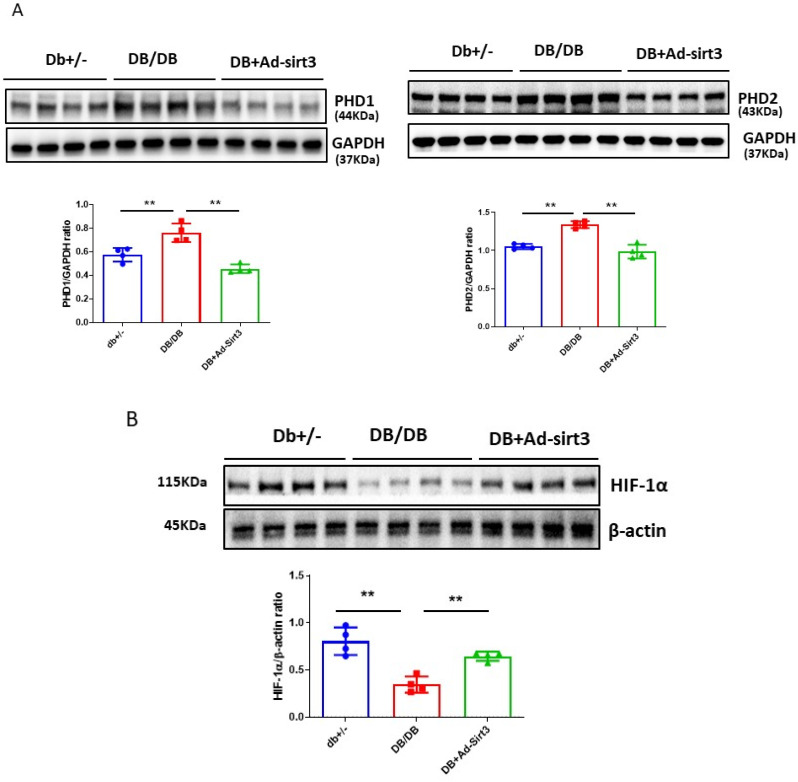
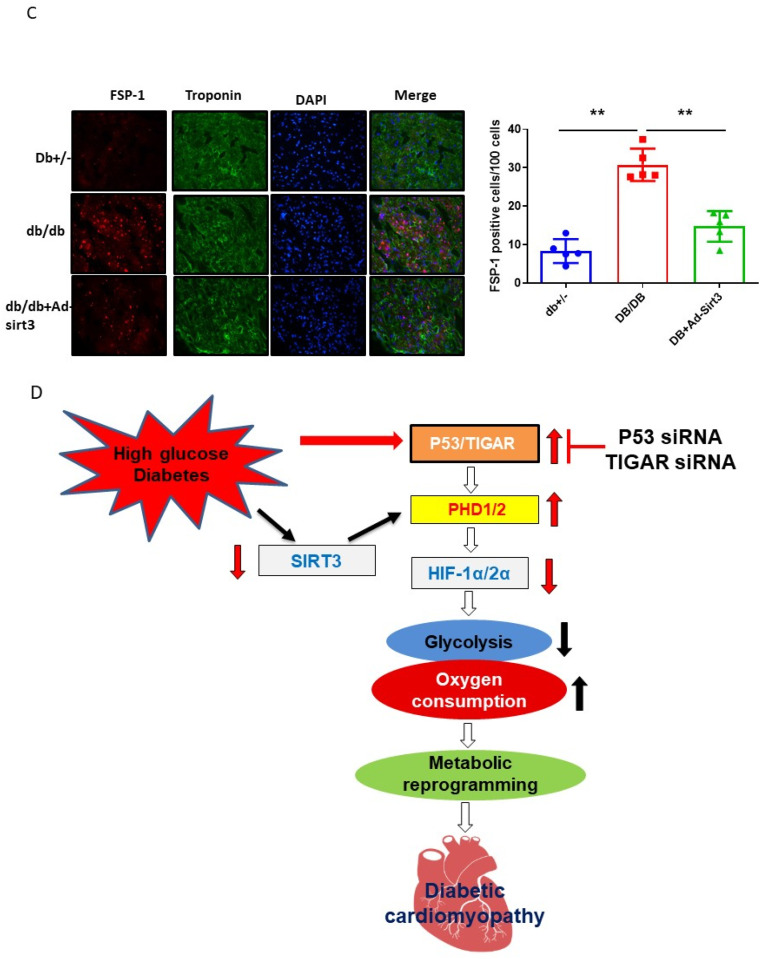
The induction of hypoxia tolerance has emerged as a novel therapeutic strategy for the treatment of ischemic diseases. The disruption of hypoxic signaling by hyperglycemia has been shown to contribute to diabetic cardiomyopathy. In this study, we explored the potential molecular mechanisms by which high glucose (HG) impairs hypoxia-inducible factor-α (HIF-α) signaling in cardiomyocytes. The exposure of H9c2 cell lines to HG resulted in time- and concentration-dependent decreases in HIF-1α and HIF-2α expression together with an increase in prolyl hydroxylase-1,2 (PHD1 and PHD2) expression, the main regulators of HIF-α destabilization in the heart. The exposure of H9c2 cells to normal glucose (5.5 mM) and high glucose (15, 30, and 45 mM) led to dose-dependent increases in p53 and TIGAR and a decrease in SIRT3 expression. The pretreatment of H9c2 with p53 siRNA to knockdown p53 attenuated PHD1 and PHD2 expression, thus significantly enhancing HIF-1α and HIF-2α expression in H9c2 cells under HG conditions. Interestingly, pretreatment with p53 siRNA altered H9c2 cell metabolism by reducing oxygen consumption rate and increasing glycolysis. Similarly, pretreatment with TIGAR siRNA blunted HG-induced PHD1 and PHD2 expression. This was accompanied by an increase in HIF-1α and HIF-2α expression with a reduction in oxygen consumption rate in H9c2 cells. Furthermore, pretreatment with adenovirus-SIRT3 (Ad-SIRT3) significantly reduced the HG-induced expression of p53 and PHDs and increased HIF-1α levels in H9c2 cells. Ad-SIRT3 treatment also regulated PHDs-HIF-1α levels in the hearts of diabetic db/db mice. Our study revealed a novel role of the HG-induced disruption of PHDs-HIF-α signaling via upregulating p53 and TIGAR expression. Therefore, the p53/TIGAR signaling pathway may be a novel target for diabetic cardiomyopathy.
Keywords: HIF-1α; TIGAR; glycolysis; high glucose; p53; prolyl hydroxylases (PHDs).
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Sirtuin 3 Alleviates Diabetic Cardiomyopathy by Regulating TIGAR and Cardiomyocyte Metabolism.J Am Heart Assoc. 2021 Feb;10(5):e018913. doi: 10.1161/JAHA.120.018913. Epub 2021 Feb 15. J Am Heart Assoc. 2021. PMID: 33586458 Free PMC article.
-
Expression of prolyl hydroxylases (PHDs) is selectively controlled by HIF-1 and HIF-2 proteins in nucleus pulposus cells of the intervertebral disc: distinct roles of PHD2 and PHD3 proteins in controlling HIF-1α activity in hypoxia.J Biol Chem. 2012 May 11;287(20):16975-86. doi: 10.1074/jbc.M111.334466. Epub 2012 Mar 26. J Biol Chem. 2012. PMID: 22451659 Free PMC article.
-
Propofol Alleviates Apoptosis Induced by Chronic High Glucose Exposure via Regulation of HIF-1α in H9c2 Cells.Oxid Med Cell Longev. 2019 Apr 10;2019:4824035. doi: 10.1155/2019/4824035. eCollection 2019. Oxid Med Cell Longev. 2019. PMID: 31093315 Free PMC article.
-
HIF-prolyl hydroxylases and cardiovascular diseases.Toxicol Mech Methods. 2012 Jun;22(5):347-58. doi: 10.3109/15376516.2012.673088. Toxicol Mech Methods. 2012. PMID: 22424133 Review.
-
The possible mechanisms underlying the impairment of HIF-1α pathway signaling in hyperglycemia and the beneficial effects of certain therapies.Int J Med Sci. 2013 Aug 22;10(10):1412-21. doi: 10.7150/ijms.5630. eCollection 2013. Int J Med Sci. 2013. PMID: 23983604 Free PMC article. Review.
Cited by
-
Metabolic reprogramming in cancer and senescence.MedComm (2020). 2025 Mar 4;6(3):e70055. doi: 10.1002/mco2.70055. eCollection 2025 Mar. MedComm (2020). 2025. PMID: 40046406 Free PMC article. Review.
-
Norepinephrine exacerbates LPS-induced cardiomyopathy via SIRT3/HO-1 axis-mediated ferroptosis.Crit Care. 2025 Aug 13;29(1):354. doi: 10.1186/s13054-025-05602-5. Crit Care. 2025. PMID: 40804419 Free PMC article.
References
-
- Di Filippo C., Marfella R., Cuzzocrea S., Piegari E., Petronella P., Giugliano D., Rossi F., D’Amico M. Hyper-glycemia in streptozotocin-induced diabetic rat increases infarct size associated with low levels of myocardial HO-1 during ischemia/reperfusion. Diabetes. 2005;54:803–810. doi: 10.2337/diabetes.54.3.803. - DOI - PubMed
-
- Frantz S., Calvillo L., Tillmanns J., Elbing I., Dienesch C., Bischoff H., Ertl G., Bauersachs J. Repetitive post-prandial hyperglycemia increases cardiac ischemia/reperfusion injury: Prevention by the alpha-glucosidase inhibitor acarbose. FASEB J. 2005;19:591–593. doi: 10.1096/fj.04-2459fje. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
