

Epilepsy in Dravet Syndrome-Current and Future Therapeutic Opportunities
- PMID: 37048615
- PMCID: PMC10094968
- DOI: 10.3390/jcm12072532
Epilepsy in Dravet Syndrome-Current and Future Therapeutic Opportunities
Abstract
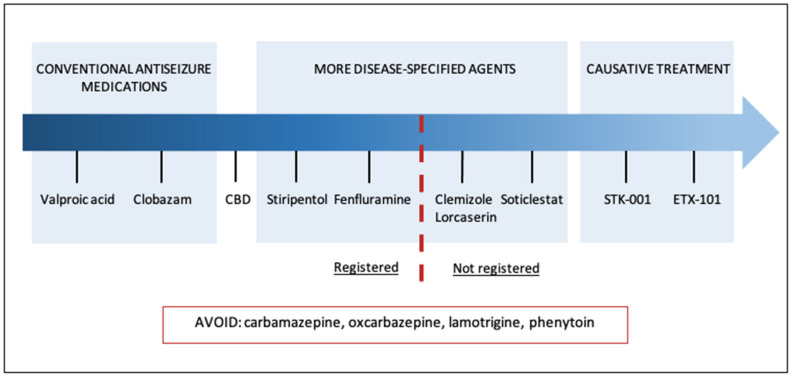
Dravet Syndrome (DS) is a developmental epileptic encephalopathy characterized by drug-resistant seizures and other clinical features, including intellectual disability and behavioral, sleep, and gait problems. The pathogenesis is strongly connected to voltage-gated sodium channel dysfunction. The current consensus of seizure management in DS consists of a combination of conventional and recently approved drugs such as stiripentol, cannabidiol, and fenfluramine. Despite promising results in randomized clinical trials and extension studies, the prognosis of the developmental outcomes of patients with DS remains unfavorable. The article summarizes recent changes in the therapeutic approach to DS and discusses ongoing clinical research directions. Serotonergic agents under investigation show promising results and may replace less DS-specific medicines. The use of antisense nucleotides and gene therapy is focused not only on symptom relief but primarily addresses the underlying cause of the syndrome. Novel compounds, after expected safe and successful implementation in clinical practice, will open a new era for patients with DS. The main goal of causative treatment is to modify the natural course of the disease and provide the best neurodevelopmental outcome with minimum neurological deficit.
Keywords: Dravet syndrome; efficacy and safety; epilepsy; novel therapeutic agents; therapeutic recommendations.
Conflict of interest statement
S.J. received honoraria from JAZZ Pharmaceuticals for participation in advisory boards. The remaining authors declare no conflict of interest.
Figures
Similar articles
-
Changing Landscape of Dravet Syndrome Management: An Overview.Neuropediatrics. 2020 Apr;51(2):135-145. doi: 10.1055/s-0040-1701694. Epub 2020 Feb 20. Neuropediatrics. 2020. PMID: 32079034 Review.
-
Comparative short-term efficacy and safety of add-on anti-seizure medications in Dravet syndrome: An indirect treatment comparison.Seizure. 2021 Oct;91:316-324. doi: 10.1016/j.seizure.2021.06.020. Epub 2021 Jun 29. Seizure. 2021. PMID: 34274891 Review.
-
A critical evaluation of fenfluramine hydrochloride for the treatment of Dravet syndrome.Expert Rev Neurother. 2022 May;22(5):351-364. doi: 10.1080/14737175.2021.1877540. Epub 2021 Feb 26. Expert Rev Neurother. 2022. PMID: 33455486
-
Therapeutic advances in Dravet syndrome: a targeted literature review.Expert Rev Neurother. 2020 Oct;20(10):1065-1079. doi: 10.1080/14737175.2020.1801423. Epub 2020 Aug 16. Expert Rev Neurother. 2020. PMID: 32799683 Review.
-
Advancements in Dravet Syndrome Therapeutics: A Comprehensive Look at Present and Future Treatment Horizons: A Focused Review.Ann Indian Acad Neurol. 2024 Jul 1;27(4):352-357. doi: 10.4103/aian.aian_49_24. Epub 2024 Aug 16. Ann Indian Acad Neurol. 2024. PMID: 39196806 Free PMC article.
Cited by
-
The role of lactate in cardiovascular diseases.Cell Commun Signal. 2023 Nov 3;21(1):317. doi: 10.1186/s12964-023-01350-7. Cell Commun Signal. 2023. PMID: 37924124 Free PMC article. Review.
-
Characterization of 13 Novel Genetic Variants in Genes Associated with Epilepsy: Implications for Targeted Therapeutic Strategies.Mol Diagn Ther. 2024 Sep;28(5):645-663. doi: 10.1007/s40291-024-00720-2. Epub 2024 Jul 14. Mol Diagn Ther. 2024. PMID: 39003674 Free PMC article.
-
The Therapeutic Role of Perampanel in Treating Pediatric Patients With Dravet Syndrome: A Scoping Review.Cureus. 2024 Jul 20;16(7):e65017. doi: 10.7759/cureus.65017. eCollection 2024 Jul. Cureus. 2024. PMID: 39165469 Free PMC article.
-
Utilizing an acute hyperthermia-induced seizure test and pharmacokinetic studies to establish optimal dosing regimens in a mouse model of Dravet syndrome.Epilepsia. 2024 Oct;65(10):3100-3114. doi: 10.1111/epi.18104. Epub 2024 Aug 30. Epilepsia. 2024. PMID: 39212337
-
Perception of psychosocial burden in mothers of children with rare pediatric neurological diseases.Sci Rep. 2025 Feb 21;15(1):6295. doi: 10.1038/s41598-025-87251-w. Sci Rep. 2025. PMID: 39984547 Free PMC article.
References
-
- Symonds J., Zuberi S.M., Stewart K., McLellan A., O’Regan M., MacLeod S., Jollands A., Joss S., Kirkpatrick M., Brunklaus A., et al. Incidence and phenotypes of childhood-onset genetic epilepsies: A prospective population-based national cohort. Brain. 2019;142:2303–2318. doi: 10.1093/brain/awz195. - DOI - PMC - PubMed
Publication types
Grants and funding
- PPN/BCN/2019/1/00066/U/00001/National Agency of Academic Exchange
- G2021026025L/International Cooperation Project of the Ministry of Science and Technology of China
- YJZX202205/Open Project of Henan Clinical Medical Research Center of Childhood Diseases
- 20-21ZY1072/Special Key Project of Henan Province Traditional Chinese Medicine Scientific Research
LinkOut - more resources
Full Text Sources
