

A General Picture of Cucurbit[8]uril Host-Guest Binding: Recalibrating Bonded Interactions
- PMID: 37049887
- PMCID: PMC10095826
- DOI: 10.3390/molecules28073124
A General Picture of Cucurbit[8]uril Host-Guest Binding: Recalibrating Bonded Interactions
Abstract
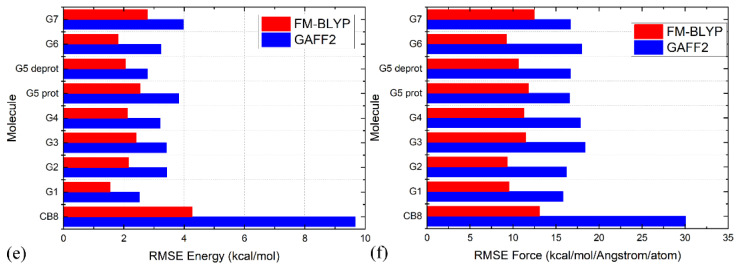
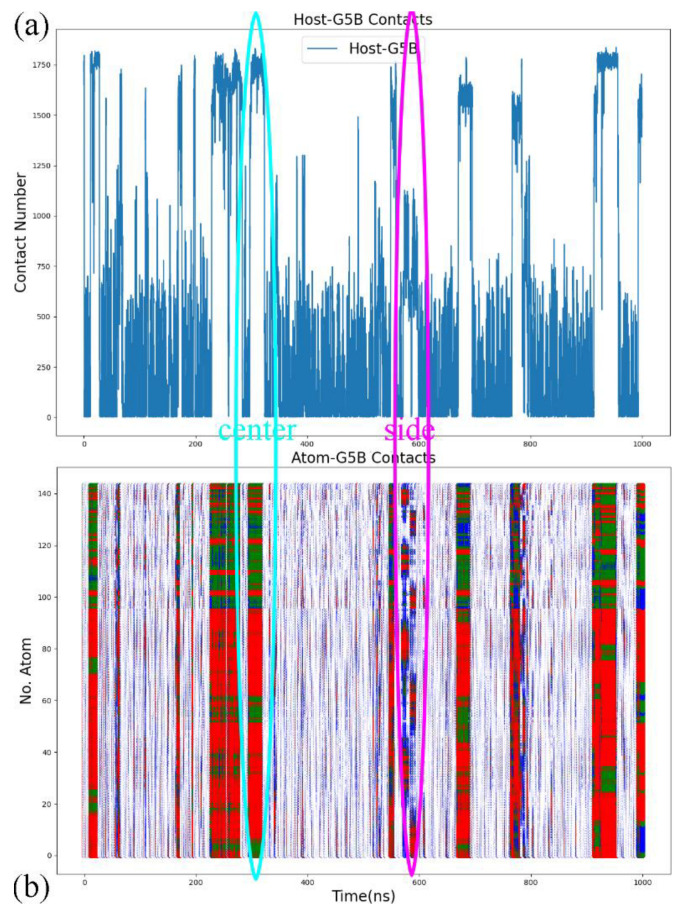
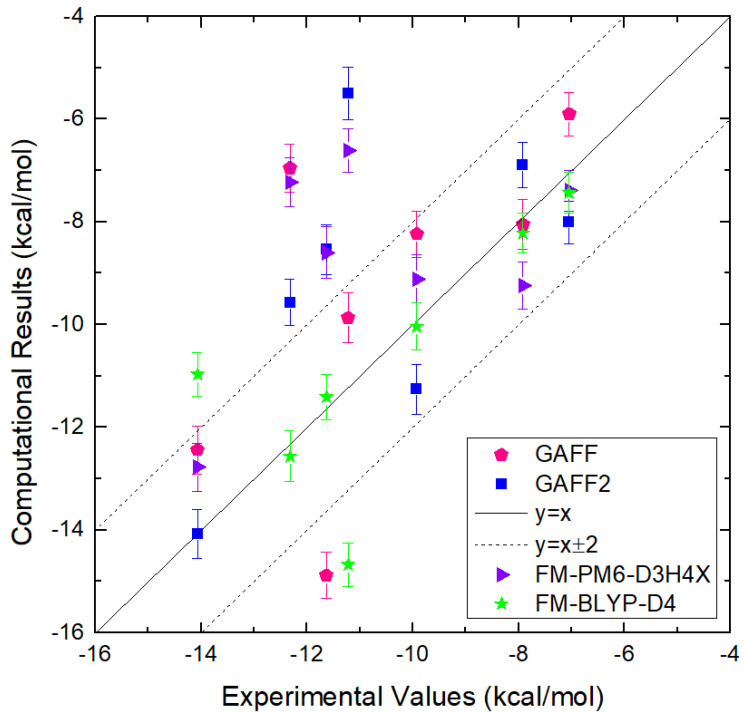
Atomic-level understanding of the dynamic feature of host-guest interactions remains a central challenge in supramolecular chemistry. The remarkable guest binding behavior of the Cucurbiturils family of supramolecular containers makes them promising drug carriers. Among Cucurbit[n]urils, Cucurbit[8]uril (CB8) has an intermediate portal size and cavity volume. It can exploit almost all host-guest recognition motifs formed by this host family. In our previous work, an extensive computational investigation of the binding of seven commonly abused and structurally diverse drugs to the CB8 host was performed, and a general dynamic binding picture of CB8-guest interactions was obtained. Further, two widely used fixed-charge models for drug-like molecules were investigated and compared in great detail, aiming at providing guidelines in choosing an appropriate charge scheme in host-guest modelling. Iterative refitting of atomic charges leads to improved binding thermodynamics and the best root-mean-squared deviation from the experimental reference is 2.6 kcal/mol. In this work, we focus on a thorough evaluation of the remaining parts of classical force fields, i.e., the bonded interactions. The widely used general Amber force fields are assessed and refitted with generalized force-matching to improve the intra-molecular conformational preference, and thus the description of inter-molecular host-guest interactions. The interaction pattern and binding thermodynamics show a significant dependence on the modelling parameters. The refitted system-specific parameter set improves the consistency of the modelling results and the experimental reference significantly. Finally, combining the previous charge-scheme comparison and the current force-field refitting, we provide general guidelines for the theoretical modelling of host-guest binding.
Keywords: Cucurbit[8]uril; abused drugs; binding mode; force fields; host–guest interaction.
Conflict of interest statement
There is no conflict of interest to declare.
Figures













Similar articles
-
A General Picture of Cucurbit[8]uril Host-Guest Binding.J Chem Inf Model. 2021 Dec 27;61(12):6107-6134. doi: 10.1021/acs.jcim.1c01208. Epub 2021 Nov 24. J Chem Inf Model. 2021. PMID: 34818004
-
Overview of the SAMPL6 host-guest binding affinity prediction challenge.J Comput Aided Mol Des. 2018 Oct;32(10):937-963. doi: 10.1007/s10822-018-0170-6. Epub 2018 Nov 10. J Comput Aided Mol Des. 2018. PMID: 30415285 Free PMC article.
-
Host-Guest Interactions between Oxaliplatin and Cucurbit[7]uril/Cucurbit[7]uril Derivatives under Pseudo-Physiological Conditions.Langmuir. 2020 Feb 11;36(5):1235-1240. doi: 10.1021/acs.langmuir.9b03325. Epub 2020 Jan 27. Langmuir. 2020. PMID: 31941282
-
The SAMPL4 host-guest blind prediction challenge: an overview.J Comput Aided Mol Des. 2014 Apr;28(4):305-17. doi: 10.1007/s10822-014-9735-1. Epub 2014 Mar 6. J Comput Aided Mol Des. 2014. PMID: 24599514 Free PMC article. Review.
-
Cucurbit[8]uril-Based Polymers and Polymer Materials.Small. 2018 Nov;14(46):e1802234. doi: 10.1002/smll.201802234. Epub 2018 Aug 31. Small. 2018. PMID: 30168673 Review.
Cited by
-
Computational Modeling of Pharmaceuticals with an Emphasis on Crossing the Blood-Brain Barrier.Pharmaceuticals (Basel). 2025 Feb 6;18(2):217. doi: 10.3390/ph18020217. Pharmaceuticals (Basel). 2025. PMID: 40006031 Free PMC article. Review.
-
Conformational States of the GDP- and GTP-Bound HRAS Affected by A59E and K117R: An Exploration from Gaussian Accelerated Molecular Dynamics.Molecules. 2024 Jan 30;29(3):645. doi: 10.3390/molecules29030645. Molecules. 2024. PMID: 38338389 Free PMC article.
-
Host Dynamics under General-Purpose Force Fields.Molecules. 2023 Aug 8;28(16):5940. doi: 10.3390/molecules28165940. Molecules. 2023. PMID: 37630194 Free PMC article.
-
Identification Mechanism of BACE1 on Inhibitors Probed by Using Multiple Separate Molecular Dynamics Simulations and Comparative Calculations of Binding Free Energies.Molecules. 2023 Jun 15;28(12):4773. doi: 10.3390/molecules28124773. Molecules. 2023. PMID: 37375328 Free PMC article.
References
Grants and funding
LinkOut - more resources
Full Text Sources
