

Tracking early lung cancer metastatic dissemination in TRACERx using ctDNA
- PMID: 37055640
- PMCID: PMC7614605
- DOI: 10.1038/s41586-023-05776-4
Tracking early lung cancer metastatic dissemination in TRACERx using ctDNA
Abstract
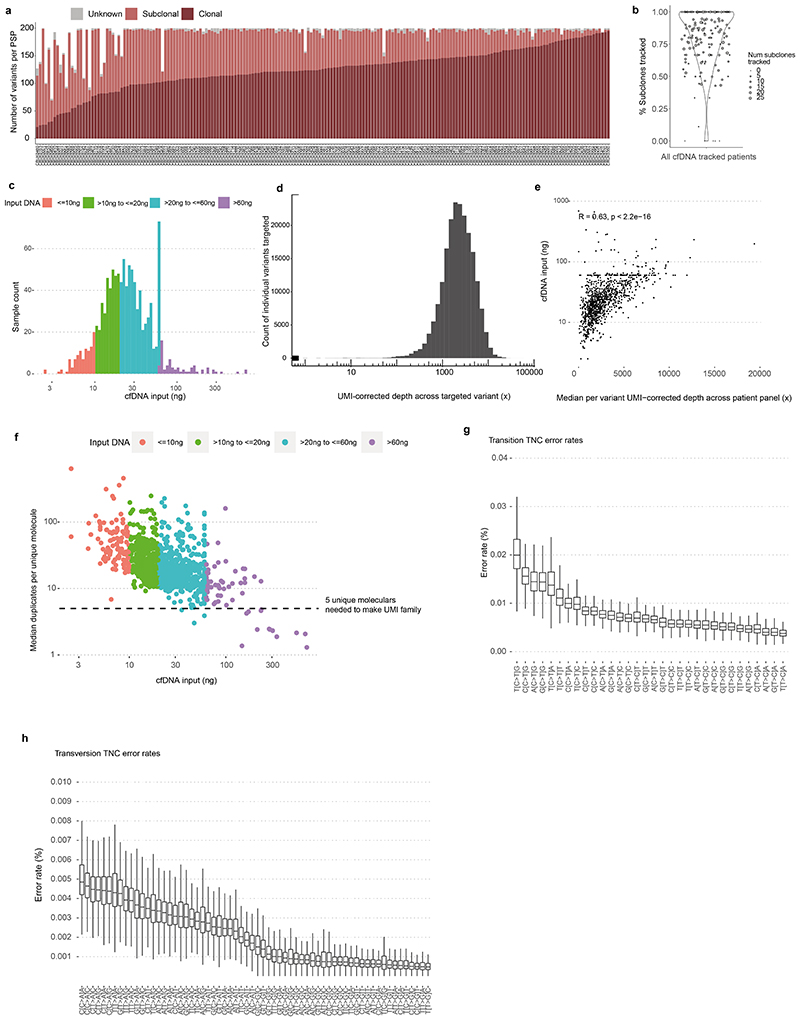
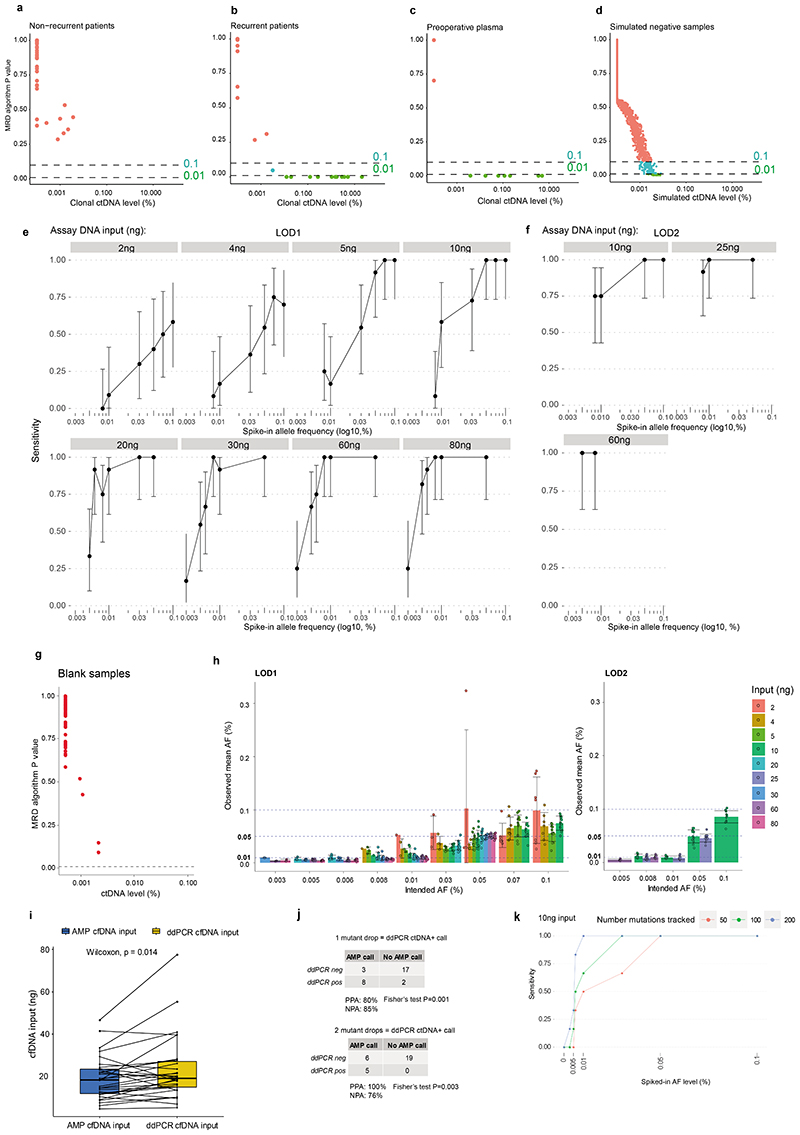
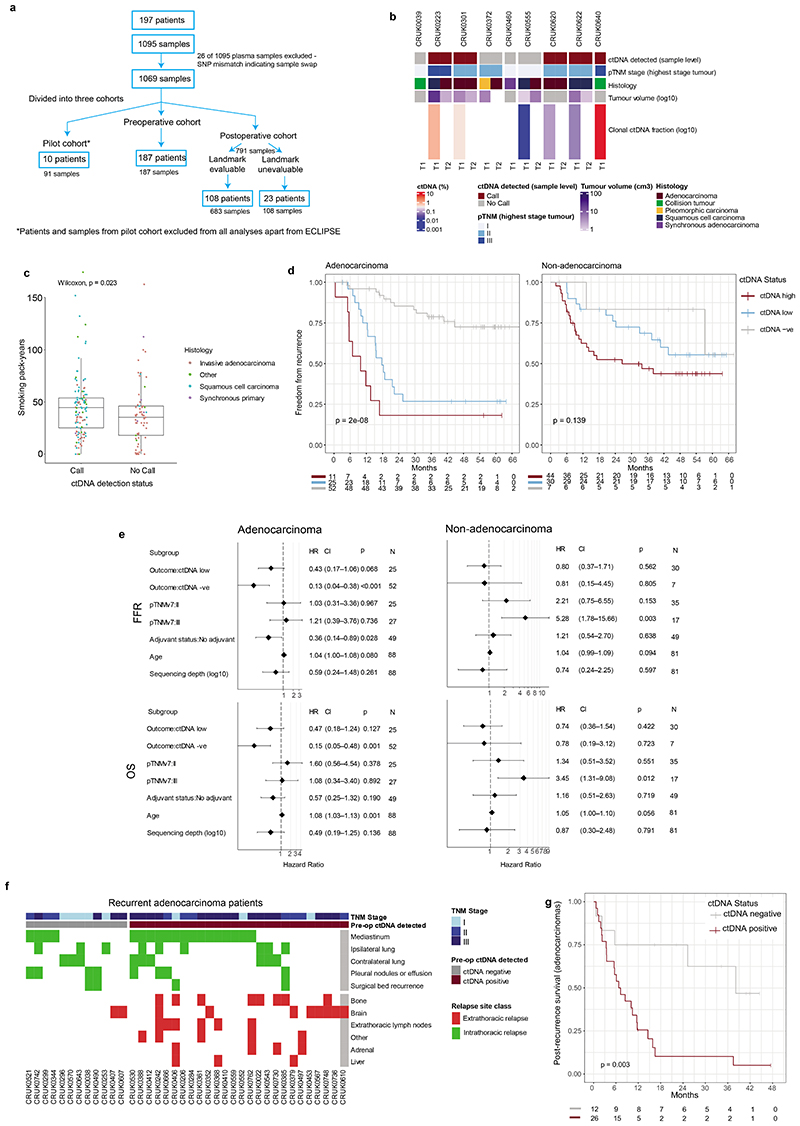
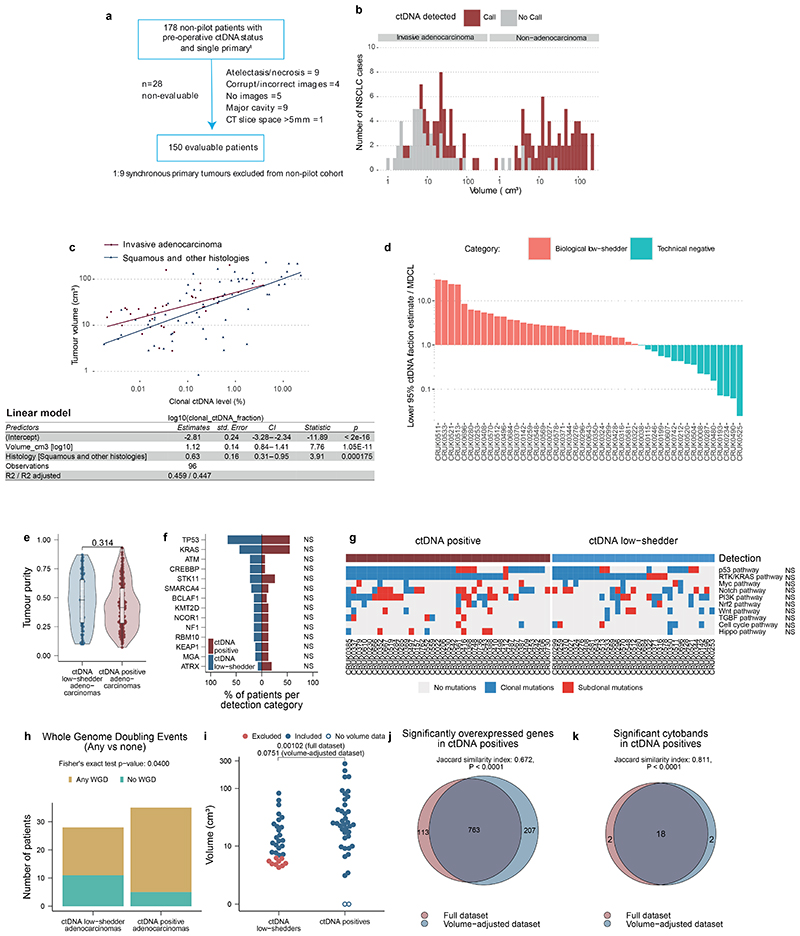
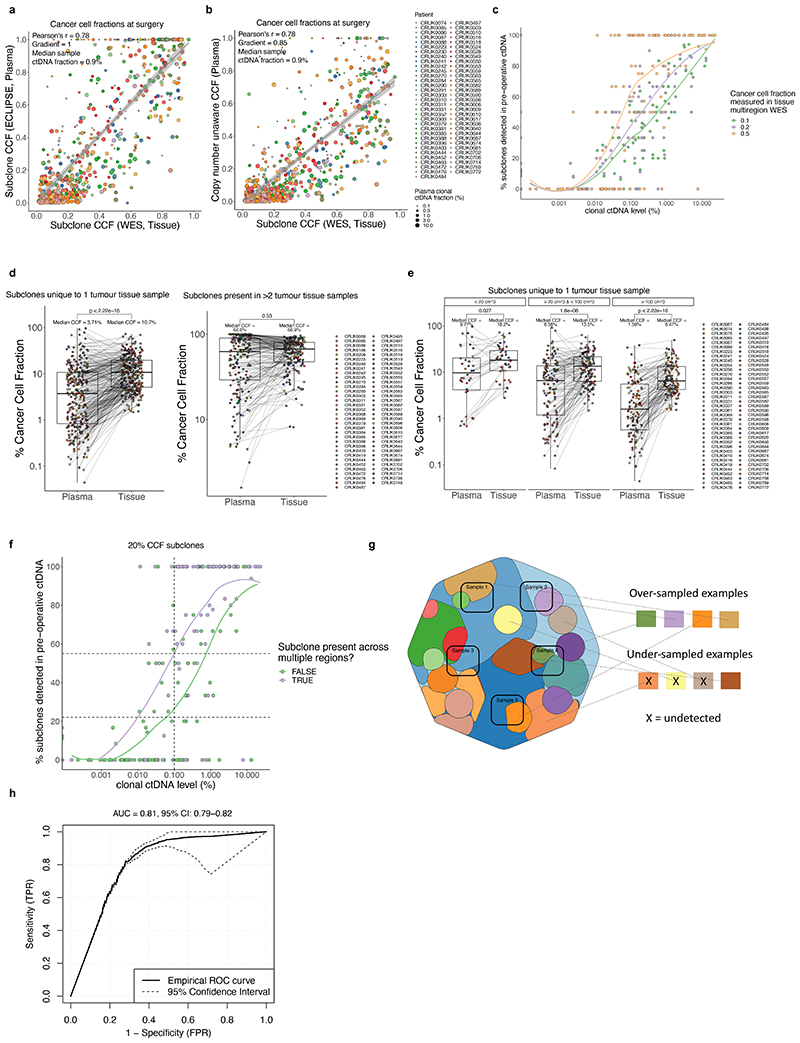
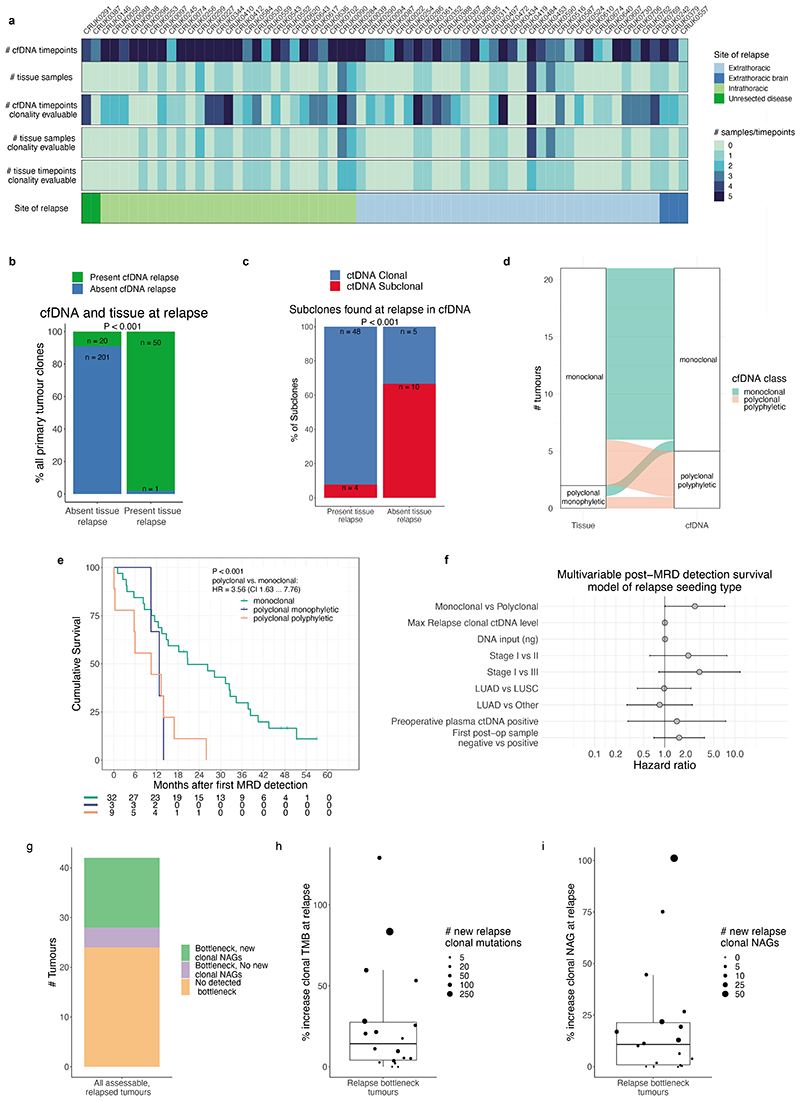
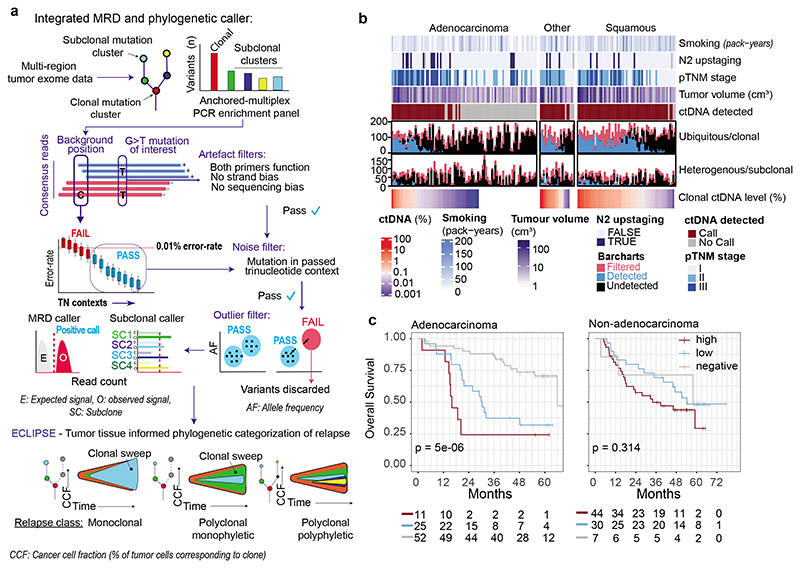
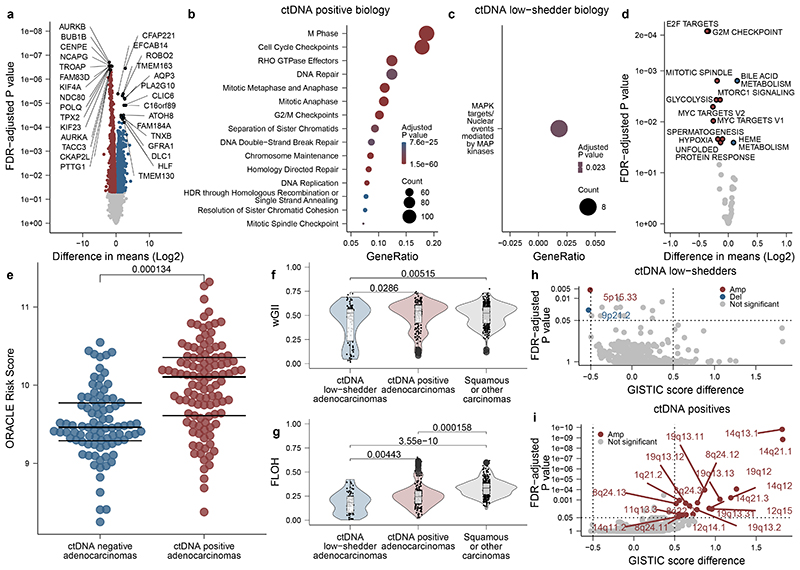
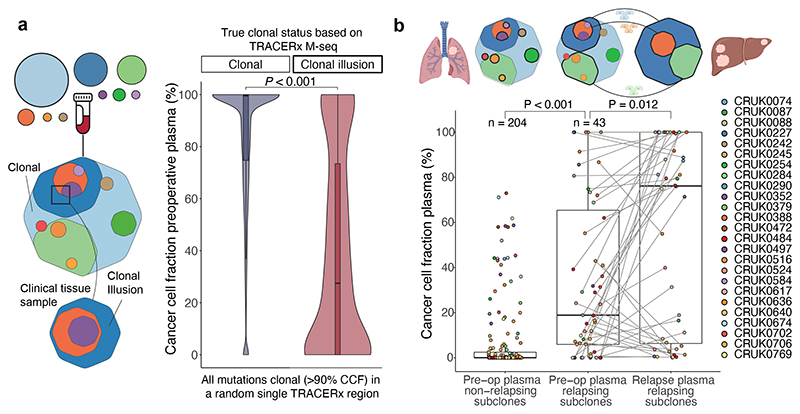
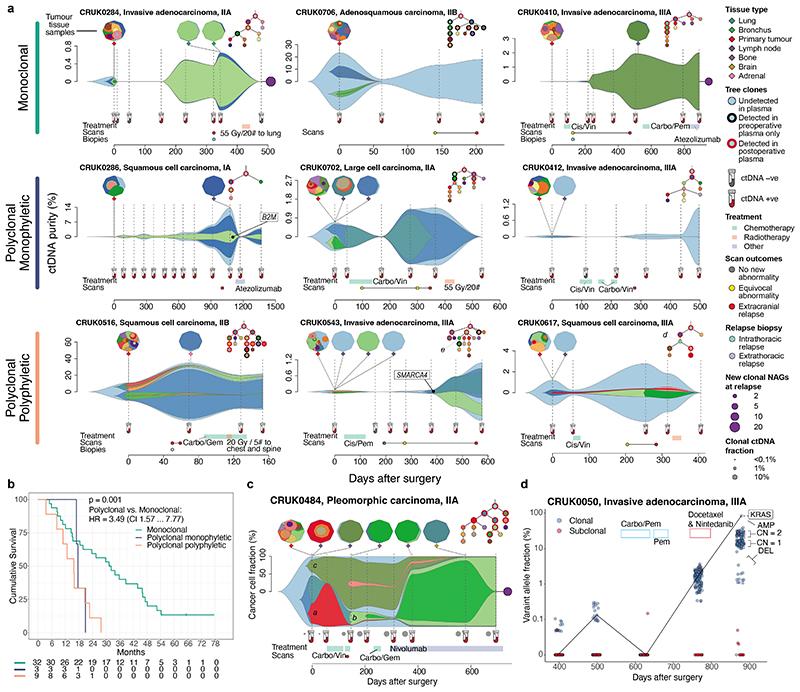
Circulating tumour DNA (ctDNA) can be used to detect and profile residual tumour cells persisting after curative intent therapy1. The study of large patient cohorts incorporating longitudinal plasma sampling and extended follow-up is required to determine the role of ctDNA as a phylogenetic biomarker of relapse in early-stage non-small-cell lung cancer (NSCLC). Here we developed ctDNA methods tracking a median of 200 mutations identified in resected NSCLC tissue across 1,069 plasma samples collected from 197 patients enrolled in the TRACERx study2. A lack of preoperative ctDNA detection distinguished biologically indolent lung adenocarcinoma with good clinical outcome. Postoperative plasma analyses were interpreted within the context of standard-of-care radiological surveillance and administration of cytotoxic adjuvant therapy. Landmark analyses of plasma samples collected within 120 days after surgery revealed ctDNA detection in 25% of patients, including 49% of all patients who experienced clinical relapse; 3 to 6 monthly ctDNA surveillance identified impending disease relapse in an additional 20% of landmark-negative patients. We developed a bioinformatic tool (ECLIPSE) for non-invasive tracking of subclonal architecture at low ctDNA levels. ECLIPSE identified patients with polyclonal metastatic dissemination, which was associated with a poor clinical outcome. By measuring subclone cancer cell fractions in preoperative plasma, we found that subclones seeding future metastases were significantly more expanded compared with non-metastatic subclones. Our findings will support (neo)adjuvant trial advances and provide insights into the process of metastatic dissemination using low-ctDNA-level liquid biopsy.
Trial registration: ClinicalTrials.gov NCT01888601.
© 2023. The Author(s).
Conflict of interest statement
C.A. has received speaking honoraria or expenses from AstraZeneca and Bristol Myers Squibb and reports employment at AstraZeneca. C.A. and C.S. are inventors on a European patent application relating to assay technology to detect tumour recurrence (PCT/GB2017/053289). This patent has been licensed to commercial entities and under their terms of employment C.A and C.S are due a revenue share of any revenue generated from such license(s). C.A. and C.S.. declare a patent application (PCT/US2017/028013) for methods to detect lung cancer. A.M.F., C.A. and C.S. are named inventors on a patent application to determine methods and systems for tumour monitoring (PCT/EP2022/077987). C.A., C.S., K.L., C.P., T.H., L.J., M.R.S, A.G., and A.L., are named inventors on a provisional patent protection related to a ctDNA detection algorithm. S.V is a co-inventor to a patent of methods for detecting molecules in a sample (U.S. Patent # 10,578,620). T.H., A.G., M.M, A.C., A.S., A.O., L.J., P.K., M.R.S, R.D..D., A.L. and J.S. are former or current employees of Invitae or ArcherDx and report stock ownership. D.B. reports personal fees from NanoString and AstraZeneca and has a patent (PCT/GB2020/050221) application on methods for cancer prognostication. M.A.B. has consulted for Achilles Therapeutics.D.A.M reports speaker fees from AstraZeneca, Eli Lilly and Takeda, consultancy fees from AstraZeneca, Thermo Fisher, Takeda, Amgen, Janssen, MIM Software, Bristol-Myers Squibb and Eli Lilly and has received educational support from Takeda and Amgen. N.G-H., A.L-H, H.B., D.H., D.S., and J.C.B., report stock ownership and employment at AstraZeneca. A.Ha. has received fees for being a member of Independent Data Monitoring Committees for Roche-sponsored clinical trials, and academic projects co-ordinated by Roche. C. T. H. has received speaker fees from AstraZeneca. M.J-H. has consulted for, and is a member of, the Achilles Therapeutics Scientific Advisory Board and Steering Committee, has received speaker honoraria from Pfizer, Astex Pharmaceuticals, Oslo Cancer Cluster, and is co-inventor on a European patent application relating to methods to detect lung cancer (PCT/US2017/028013). This patent has been licensed to commercial entities and under terms of employment M.J-H. is due a share of any revenue generated from such license(s).N.J.B. is a co-inventor to a patent to identify responders to cancer treatment (PCT/GB2018/051912), has a patent application (PCT/GB2020/050221) on methods for cancer prognostication and a patent on methods for predicting anti-cancer response (US14/466,208). H.J.W.L.A. acknowledges financial support from NIH (NIH-USA U24CA194354, NIH-USA U01CA190234, NIH-USA U01CA209414, and NIH-USA R35CA22052), and the European Union - European Research Council (HA: 866504). H.J.W.L.A. has also received personal fees and stock from Onc.AI, Sphera LLC and Love Health Inc, and speaking honoraria from Bristol Myers Squibb. K.L. has a patent on indel burden and CPI response pending and speaker fees from Roche tissue diagnostics and Ellipses Pharmaceuticals, research funding from CRUK TDL/Ono/LifeArc alliance, Genesis Therapeutics and consulting roles with Monopteros Therapeutics and Kynos Therapeutics (all outside of this work). N.M. has received consultancy fees and has stock options in Achilles Therapeutics. N.M. holds European patents relating to targeting neoantigens (PCT/EP2016/059401), identifying patient response to immune checkpoint blockade (PCT/EP2016/071471), determining HLA LOH (PCT/GB2018/052004), predicting survival rates of patients with cancer (PCT/GB2020/050221). C.S. acknowledges grant support from AstraZeneca, Boehringer-Ingelheim, Bristol Myers Squibb, Pfizer, Roche-Ventana, Invitae (previously Archer Dx Inc - collaboration in minimal residual disease sequencing technologies), Ono Pharmaceutical, and Personalis. He is an AstraZeneca Advisory Board member and Chief Investigator for the AZ MeRmaiD 1 and 2 clinical trials and is also Co-Chief Investigator of the NHS Galleri trial funded by GRAIL and a paid member of GRAIL's Scientific Advisory Board. He receives consultant fees from Achilles Therapeutics (also SAB member), Bicycle Therapeutics (also a SAB member), Genentech, Medicxi, Roche Innovation Centre – Shanghai, Metabomed (until July 2022), and the Sarah Cannon Research Institute C.S has received honoraria from Amgen, AstraZeneca, Pfizer, Novartis, GlaxoSmithKline, MSD, Bristol Myers Squibb, Illumina, and Roche-Ventana. C.S. had stock options in Apogen Biotechnologies and GRAIL until June 2021, and currently has stock options in Epic Bioscience, Bicycle Therapeutics, and has stock options and is co-founder of Achilles Therapeutics. C.S. holds additional patent applications related to targeting neoantigens (PCT/EP2016/059401), identifying patient response to immune checkpoint blockade (PCT/EP2016/071471), determining HLA LOH (PCT/GB2018/052004), predicting survival rates of patients with cancer (PCT/GB2020/050221), identifying patients who respond to cancer treatment (PCT/GB2018/051912) and both a European and US patent application related to identifying insertion/deletion mutation targets (PCT/GB2018/051892).
Figures
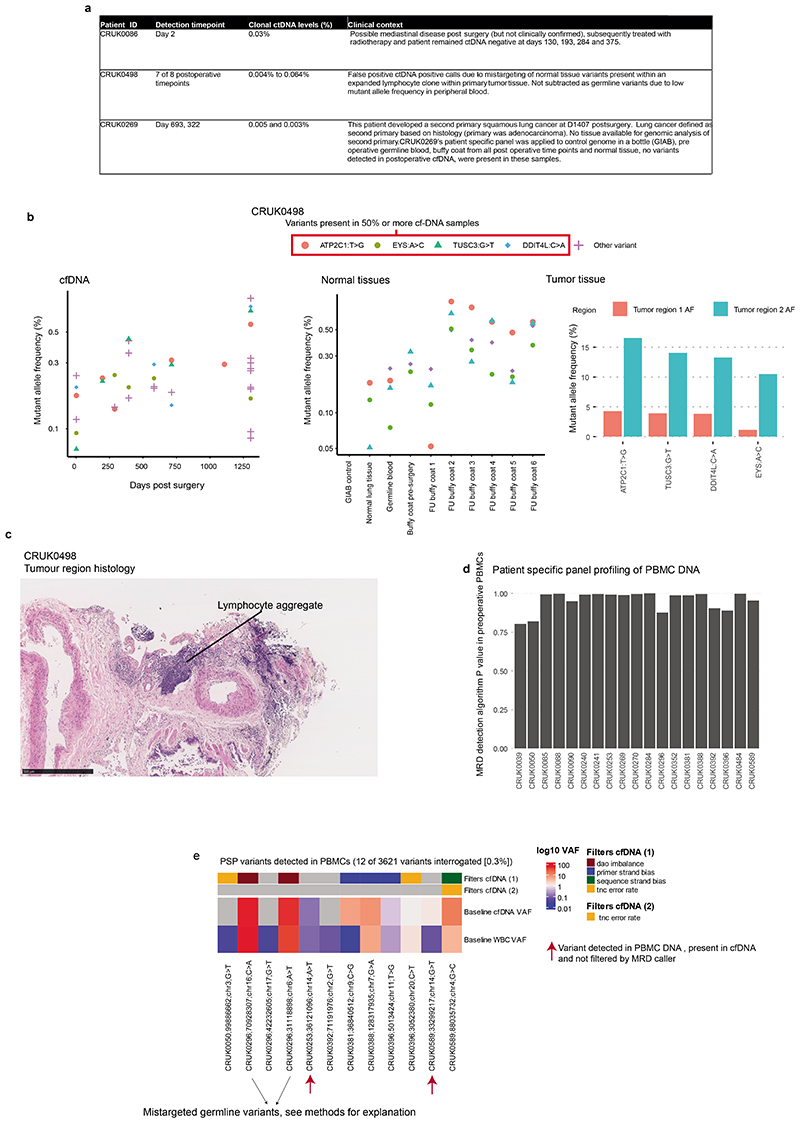
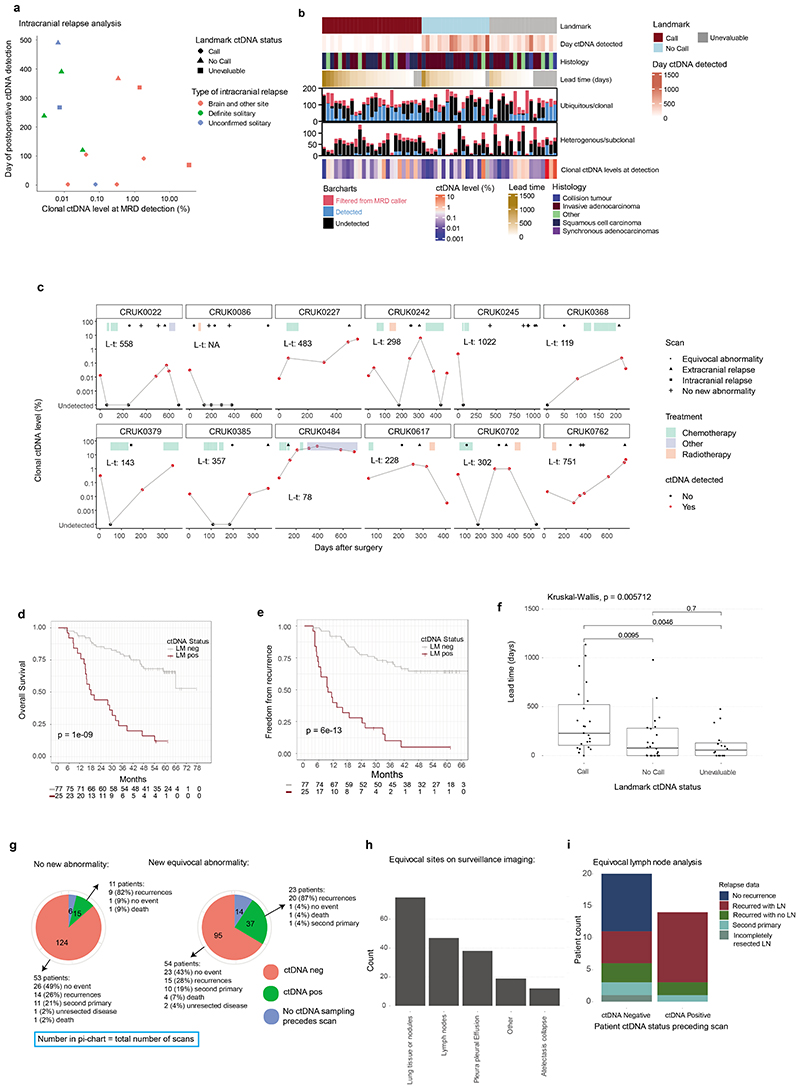
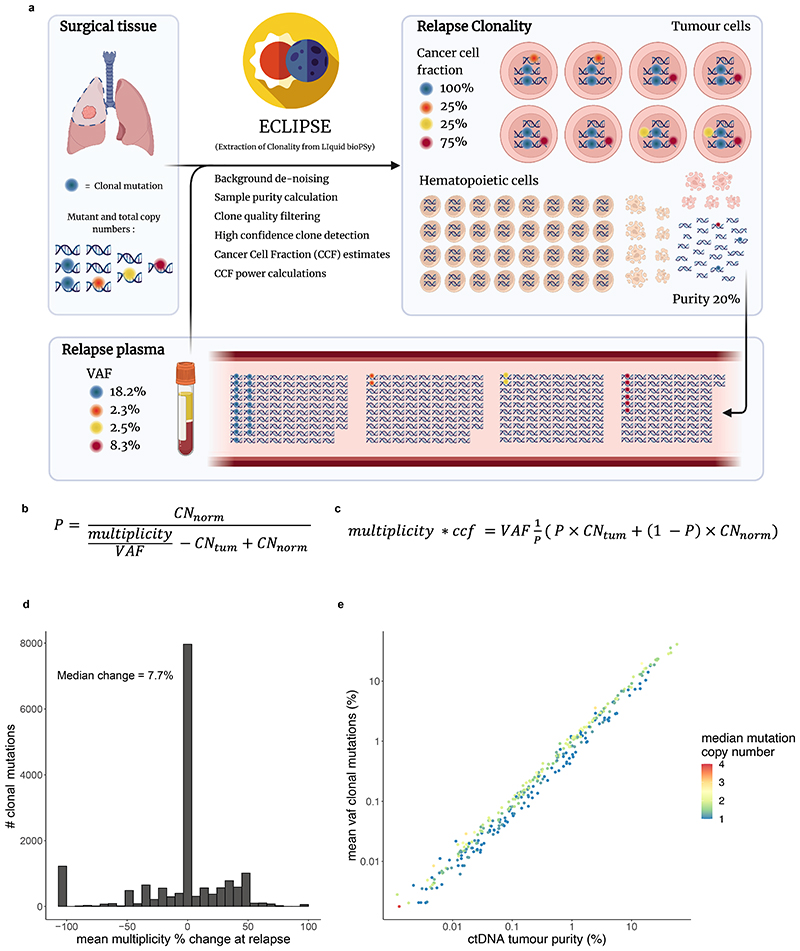
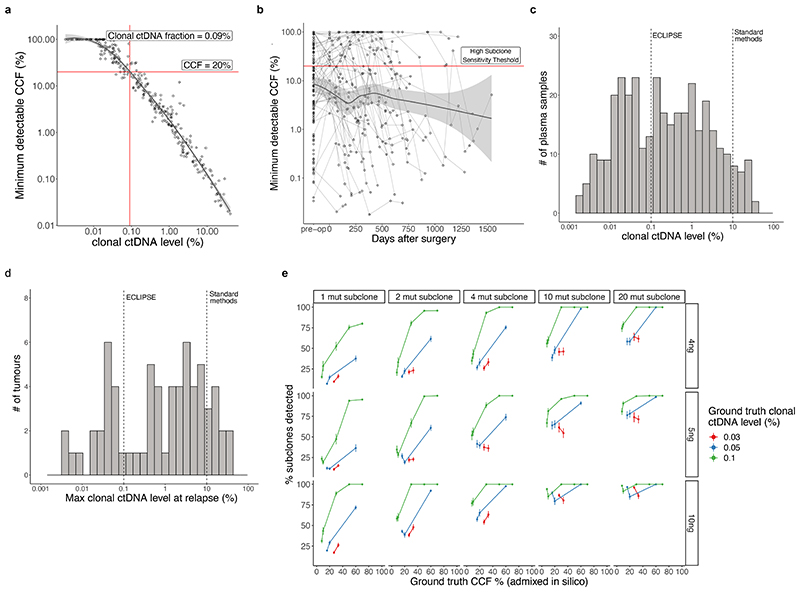

Comment in
-
Molecular portraits of lung cancer evolution.Nature. 2023 Apr;616(7957):435-436. doi: 10.1038/d41586-023-00934-0. Nature. 2023. PMID: 37045956 No abstract available.
References
-
- Moding EJ, Nabet BY, Alizadeh AA, Diehn M. Detecting Liquid Remnants of Solid Tumors: Circulating Tumor DNA Minimal Residual Disease. Cancer Discov. 2021;11:2968–2986. doi: 10.1158/2159-8290.CD-21-0634. - DOI - PMC - PubMed
-
- Jamal-Hanjani M, et al. Tracking the Evolution of Non-Small-Cell Lung Cancer. N Engl J Med. 2017;376:2109–2121. - PubMed
-
- Xia L, et al. Perioperative ctDNA-based Molecular Residual Disease Detection for Non-Small Cell Lung Cancer: A Prospective Multicenter Cohort Study (LUNGCA-1) Clin Cancer Res. 2021 - PubMed
Publication types
MeSH terms
Substances
Associated data
Grants and funding
- CTRNBC-2022/100001/CRUK_/Cancer Research UK/United Kingdom
- 30025/CRUK_/Cancer Research UK/United Kingdom
- CC2041/WT_/Wellcome Trust/United Kingdom
- CC2041/CRUK_/Cancer Research UK/United Kingdom
- U01 CA209414/CA/NCI NIH HHS/United States
- MR/V033077/1/MRC_/Medical Research Council/United Kingdom
- 29569/CRUK_/Cancer Research UK/United Kingdom
- MR/W025051/1/MRC_/Medical Research Council/United Kingdom
- 21999/CRUK_/Cancer Research UK/United Kingdom
- 24956/CRUK_/Cancer Research UK/United Kingdom
- A17786/CRUK_/Cancer Research UK/United Kingdom
- 25349/CRUK_/Cancer Research UK/United Kingdom
- 17786/CRUK_/Cancer Research UK/United Kingdom
- U01 CA190234/CA/NCI NIH HHS/United States
- U24 CA194354/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
