

A thrombophilic allele of clotting Factor VII/VIIa promoting recurrent pulmonary emboli, clinical details, and a structural model of the altered protein: a case report
- PMID: 37055848
- PMCID: PMC10100170
- DOI: 10.1186/s13256-023-03833-0
A thrombophilic allele of clotting Factor VII/VIIa promoting recurrent pulmonary emboli, clinical details, and a structural model of the altered protein: a case report
Abstract
Background: The clotting or hemostasis system is a meticulously regulated set of enzymatic reactions that occur in the blood and culminate in formation of a fibrin clot. The precisely calibrated signaling system that prevents or initiates clotting originates with the activated Factor Seven (FVIIa) complexed with tissue factor (TF) formed in the endothelium. Here we describe a rare inherited mutation in the FVII gene which is associated with pathological clotting.
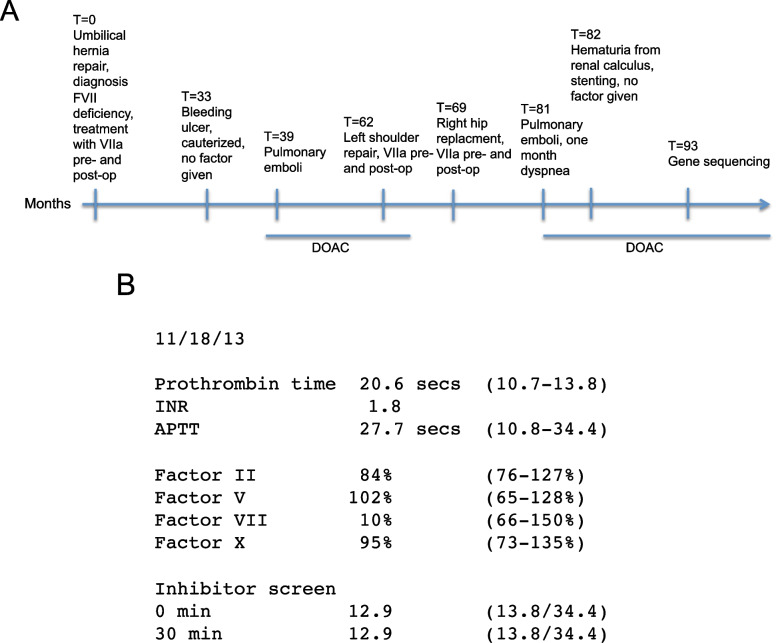
Case presentation: The 52-year-old patient, with European, Cherokee and African American origins, FS was identified as having low FVII (10%) prior to elective surgery for an umbilical hernia. He was given low doses of NovoSeven (therapeutic Factor VIIa) and had no unusual bleeding or clotting during the surgery. In fact, during his entire clinical course he had no unprovoked bleeding. Bleeding instances occurred with hemostatic stresses such as gastritis, kidney calculus, orthopedic surgery, or tooth extraction, and these were handled without factor replacement. On the other hand, FS sustained two unprovoked and life-threatening instances of pulmonary emboli, although he was not treated with NovoSeven at any time close to the events. Since 2020, he has been placed on a DOAC (Direct Oral Anticoagulant, producing Factor Xa inhibition) and has sustained no further clots.
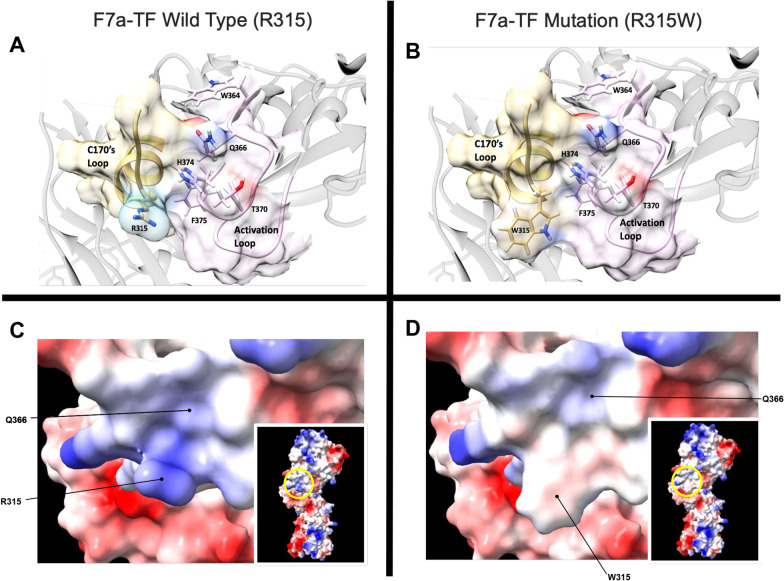
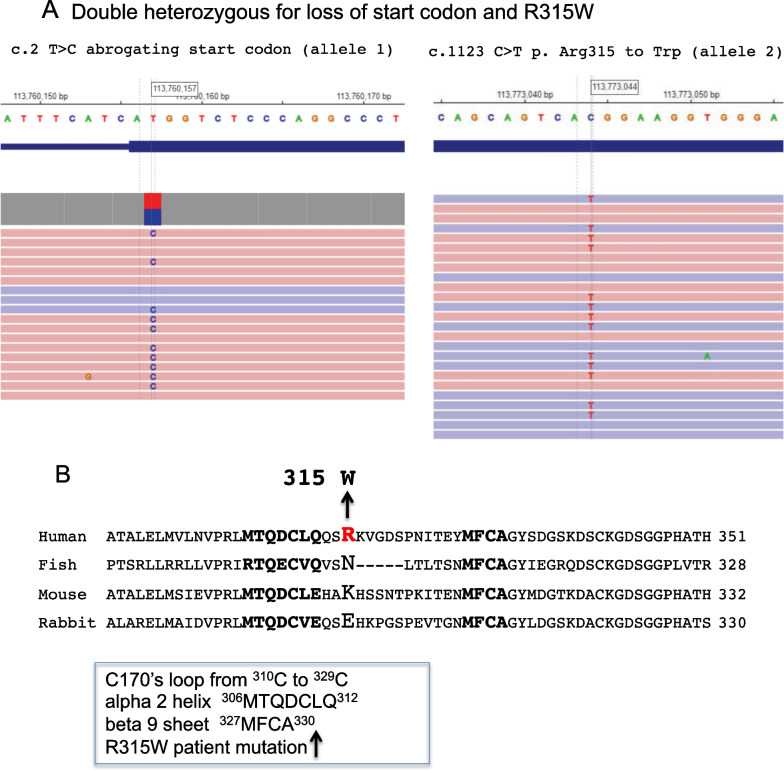
Possible mechanism of (unauthorized) fvii activation: FS has a congenitally mutated FVII/FVIIa gene, which carries a R315W missense mutation in one allele and a mutated start codon (ATG to ACG) in the other allele, thus rendering the patient effectively homozygous for the missense FVII. Structure based comparisons with known crystal structures of TF-VIIa indicate that the patient's missense mutation is predicted to induce a conformational shift of the C170's loop due to crowding of the bulky tryptophan to a distorted "out" position (Fig. 1). This mobile loop likely forms new interactions with activation loop 3, stabilizing a more active conformation of the FVII and FVIIa protein. The mutant form of FVIIa may be better able to interact with TF, displaying a modified serine protease active site with enhanced activity for downstream substrates such as Factor X.
Conclusions: Factor VII can be considered the gatekeeper of the coagulation system. Here we describe an inherited mutation in which the gatekeeper function is altered. Instead of the expected bleeding manifestations resulting from a clotting factor deficiency, the patient FS suffered clotting episodes. The efficacy of the DOAC in treating and preventing clots in this unusual situation is due to its target site of inhibition (anti-Xa), which lies downstream of the site of action of FVIIa/TF.
Keywords: DOAC; Factor VII; Factor VIIa; Hemostasis; Thrombophilia; Tissue Factor (TF); Zymogen.
© 2023. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
Similar articles
-
A frequent human coagulation Factor VII mutation (A294V, c152) in loop 140s affects the interaction with activators, tissue factor and substrates.Biochem J. 2002 Apr 15;363(Pt 2):411-6. doi: 10.1042/0264-6021:3630411. Biochem J. 2002. PMID: 11931672 Free PMC article.
-
Factor VII mutant V154G models a zymogen-like form of factor VIIa.Biochem J. 2003 Feb 1;369(Pt 3):563-71. doi: 10.1042/BJ20020888. Biochem J. 2003. PMID: 12358603 Free PMC article.
-
Inhibitors of Tissue Factor. Factor VIIa for anticoagulant therapy.Curr Med Chem. 2004 Sep;11(17):2275-90. doi: 10.2174/0929867043364568. Curr Med Chem. 2004. PMID: 15379712 Review.
-
Tissue factor pathway.Baillieres Clin Haematol. 1994 Sep;7(3):469-84. doi: 10.1016/s0950-3536(05)80094-0. Baillieres Clin Haematol. 1994. PMID: 7841596 Review.
-
The tissue factor/factor VIIa/factor Xa complex: a model built by docking and site-directed mutagenesis.Proteins. 2003 Nov 15;53(3):640-8. doi: 10.1002/prot.10445. Proteins. 2003. PMID: 14579355
References
-
- Morrissey JH, Broze GJ Jr. Tissue Factor and the Initiation and Regulation (TFPI) of coagulation, Chapter 11. Hemostasis and thrombosis 6th edn. V. J. A. Marder, WC, Bennett JS, Schulman S, White GC. Philadelphia, Lippincott Williams & Wilkins, a Wolters Kluwer business; 2013, pp 163–178.
-
- Mariani G, Herrmann FH, Schulman S, Batorova A, Wulff K, Etro D, Dolce A, Auerswald G, Astermark J, Schved JF, Ingerslev J, Bernardi F, VIIDSG International Factor. Thrombosis in inherited factor VII deficiency. J Thromb Haemost. 2003; 1(10): 2153–2158. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
