

SEQUIN is an R/Shiny framework for rapid and reproducible analysis of RNA-seq data
- PMID: 37056373
- PMCID: PMC10088091
- DOI: 10.1016/j.crmeth.2023.100420
SEQUIN is an R/Shiny framework for rapid and reproducible analysis of RNA-seq data
Abstract
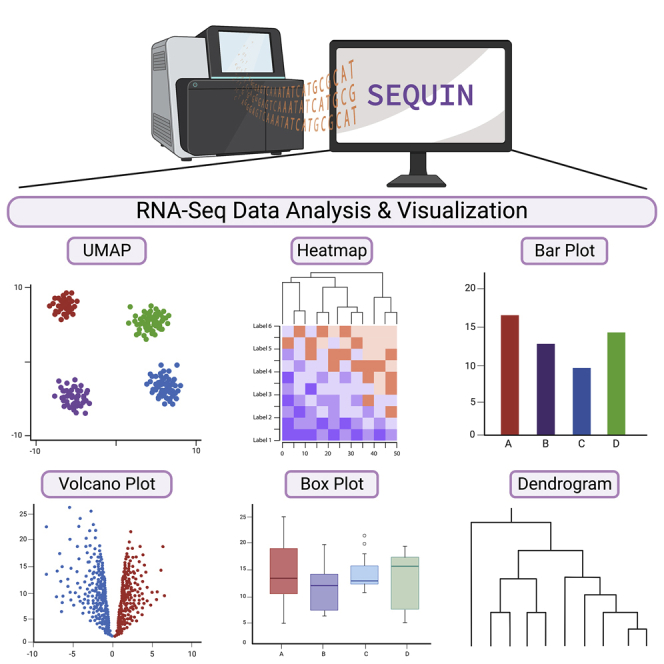
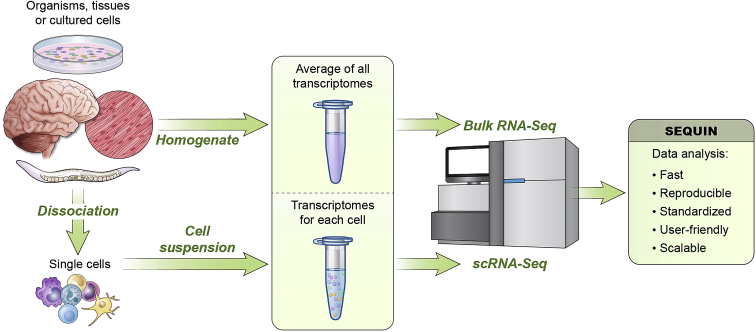
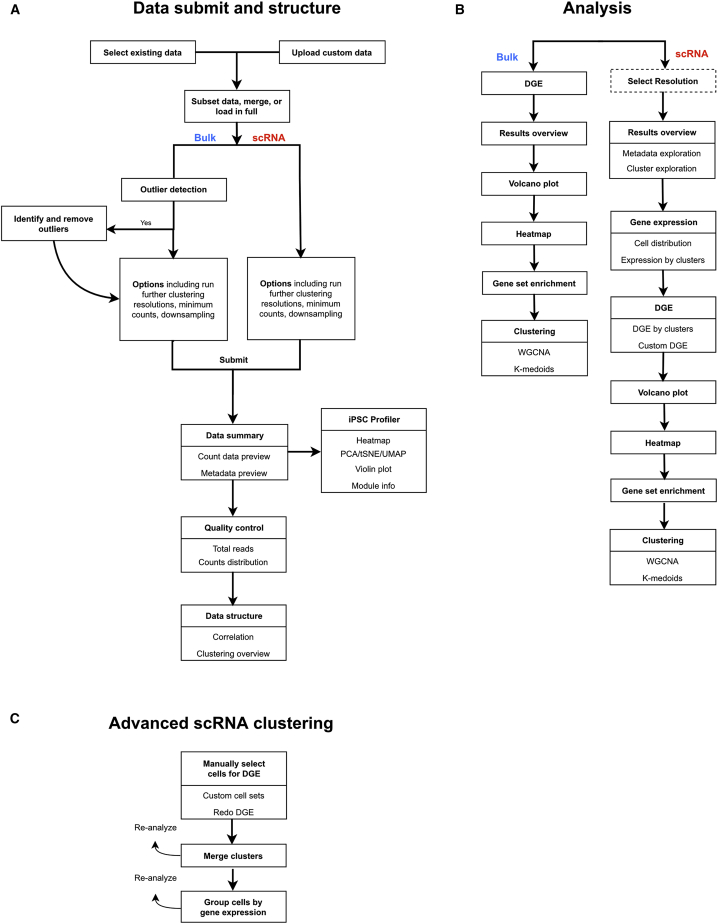
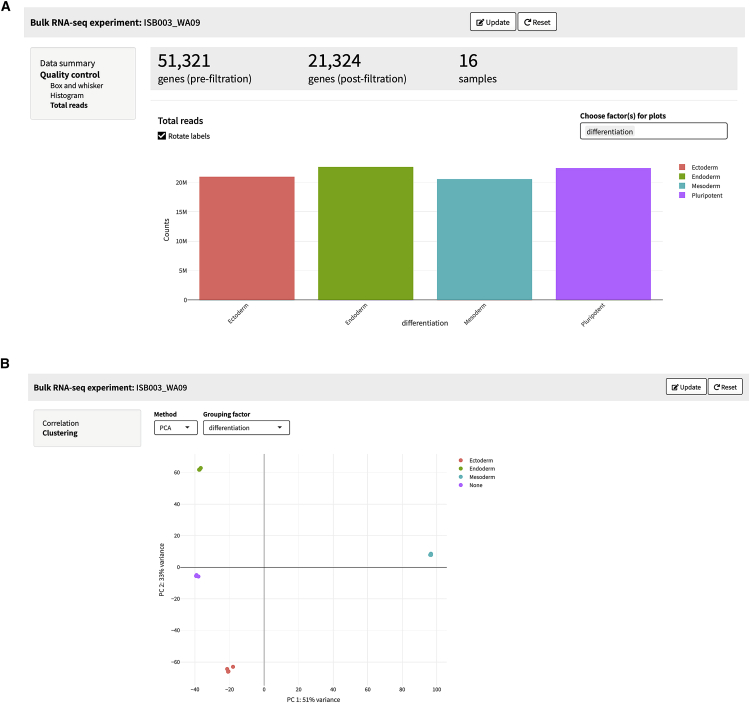
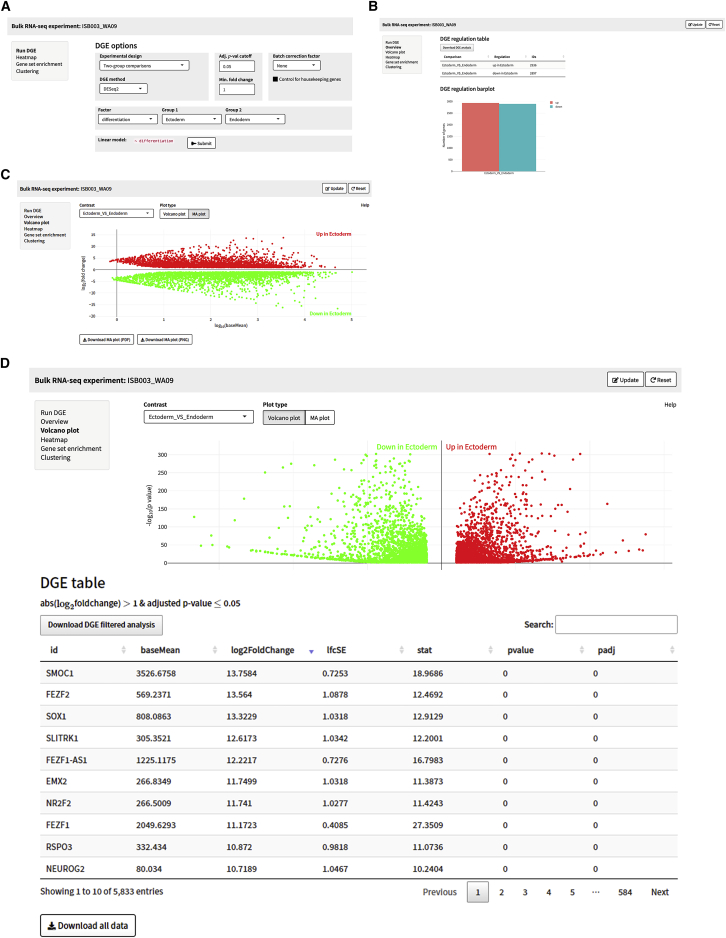
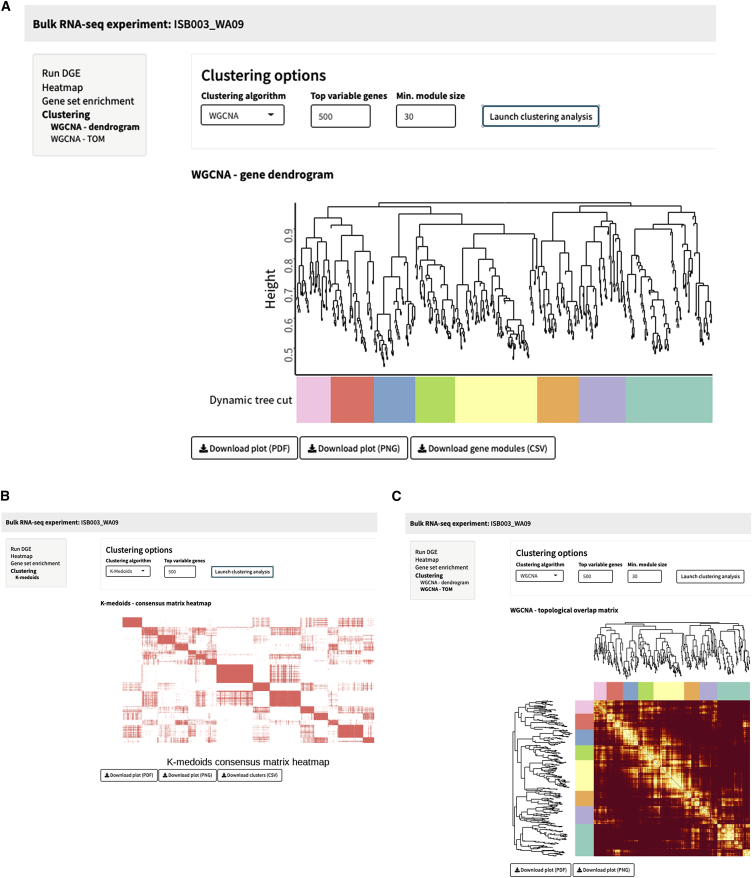
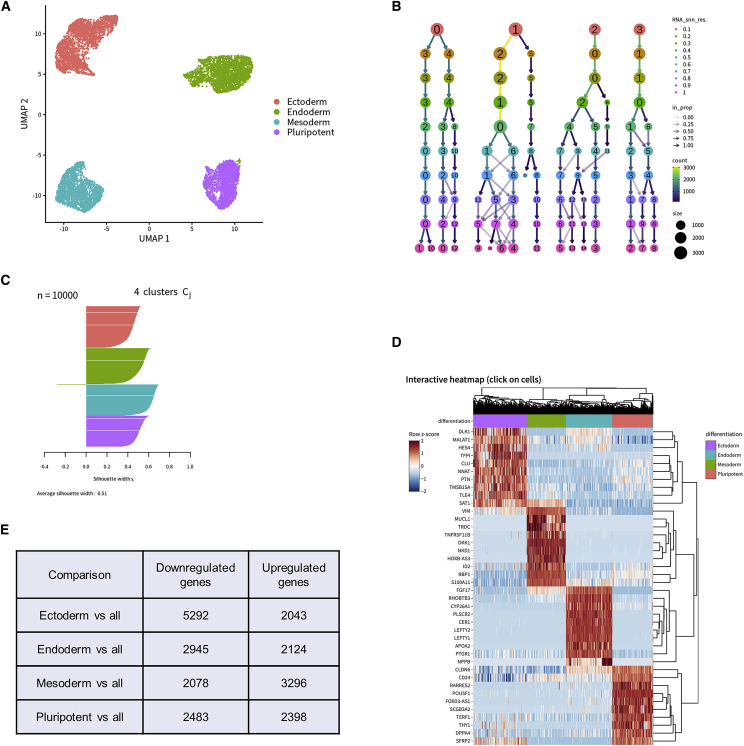
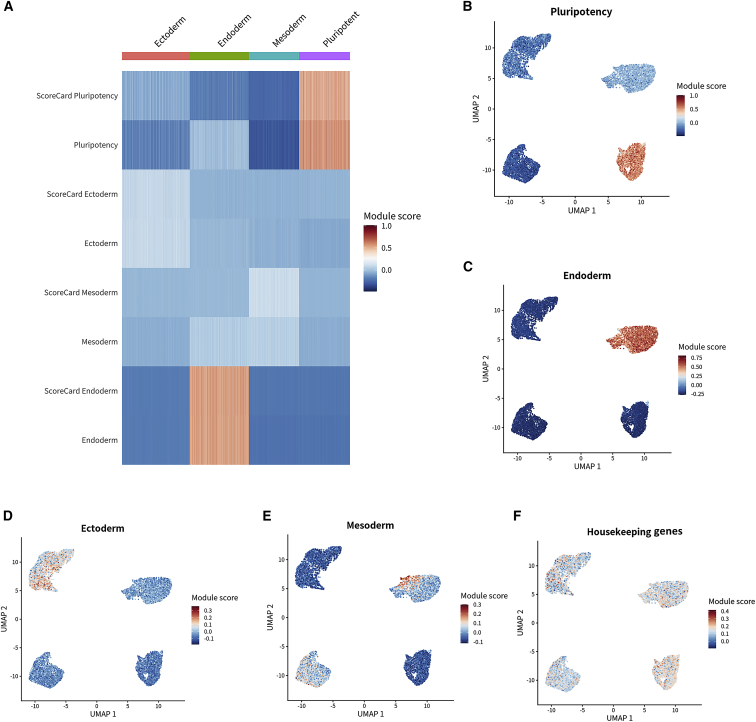
SEQUIN is a web-based application (app) that allows fast and intuitive analysis of RNA sequencing data derived for model organisms, tissues, and single cells. Integrated app functions enable uploading datasets, quality control, gene set enrichment, data visualization, and differential gene expression analysis. We also developed the iPSC Profiler, a practical gene module scoring tool that helps measure and compare pluripotent and differentiated cell types. Benchmarking to other commercial and non-commercial products underscored several advantages of SEQUIN. Freely available to the public, SEQUIN empowers scientists using interdisciplinary methods to investigate and present transcriptome data firsthand with state-of-the-art statistical methods. Hence, SEQUIN helps democratize and increase the throughput of interrogating biological questions using next-generation sequencing data with single-cell resolution.
Keywords: R/Shiny app; RNA sequencing; UMAP; data visualization; dimensionality reduction; gene expression; iPSC profiler; single-cell analysis; t-SNE; transcriptome analysis.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
References
-
- Wang Y.J., Schug J., Lin J., Wang Z., Kossenkov A., Kaestner K.H. Comparative analysis of commercially available single-cell RNA sequencing platforms for their performance in complex human tissues. bioRxiv. 2019 doi: 10.1101/541433. Preprint at. - DOI
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
