

On the limits of fitting complex models of population history to f-statistics
- PMID: 37057893
- PMCID: PMC10310323
- DOI: 10.7554/eLife.85492
On the limits of fitting complex models of population history to f-statistics
Abstract
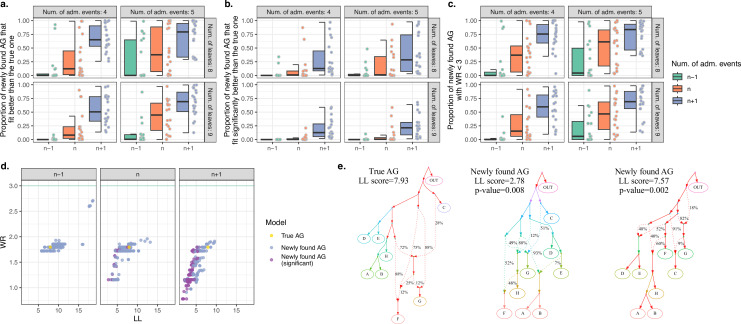
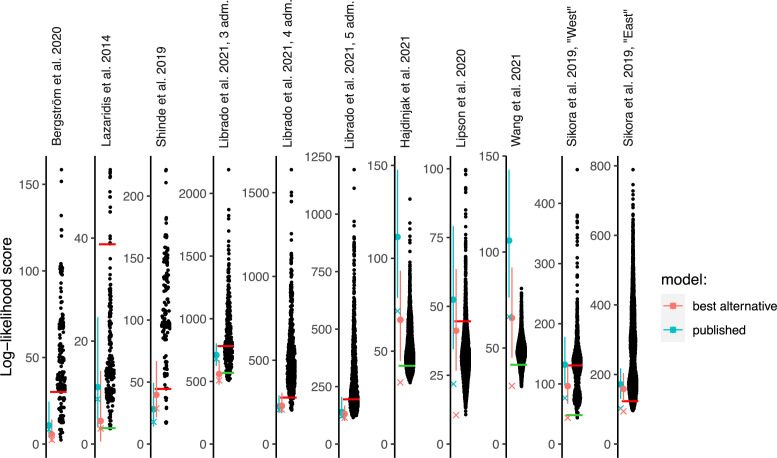
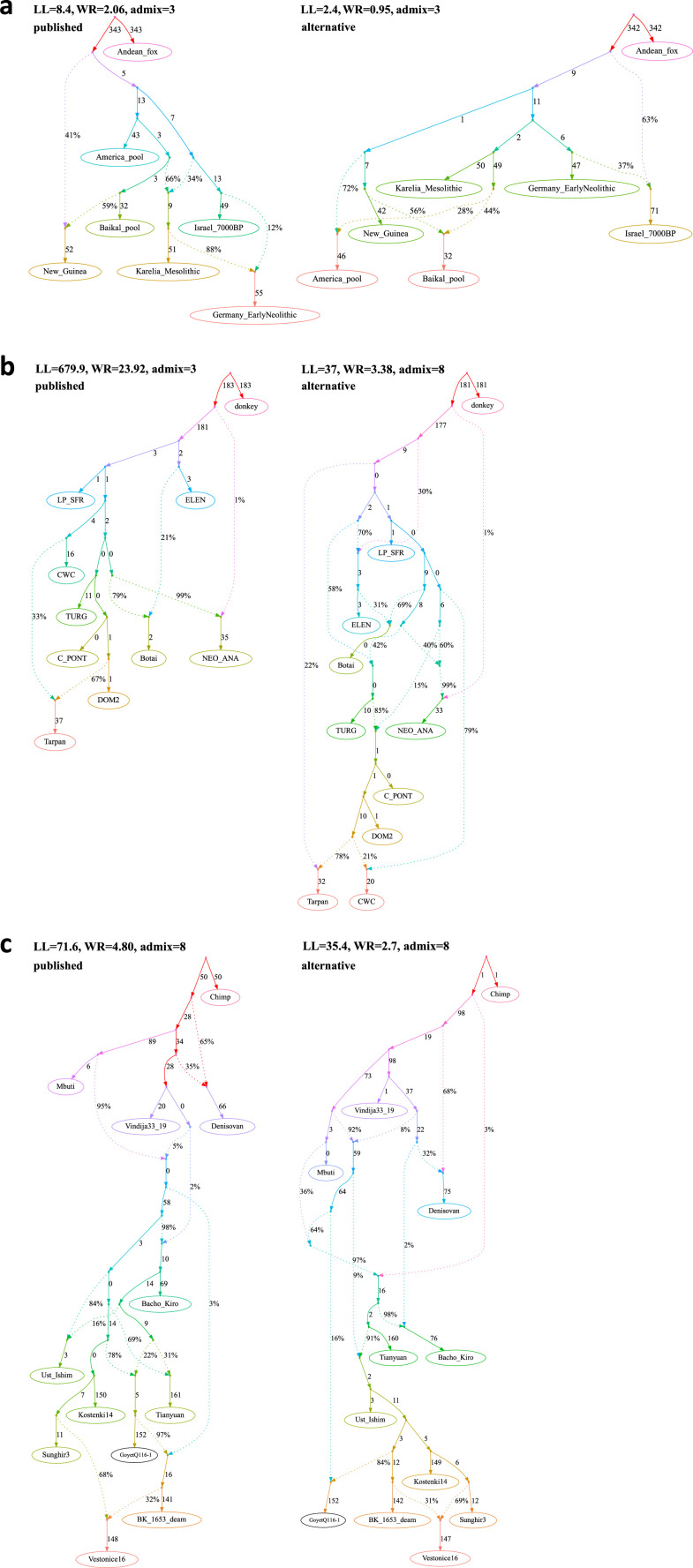
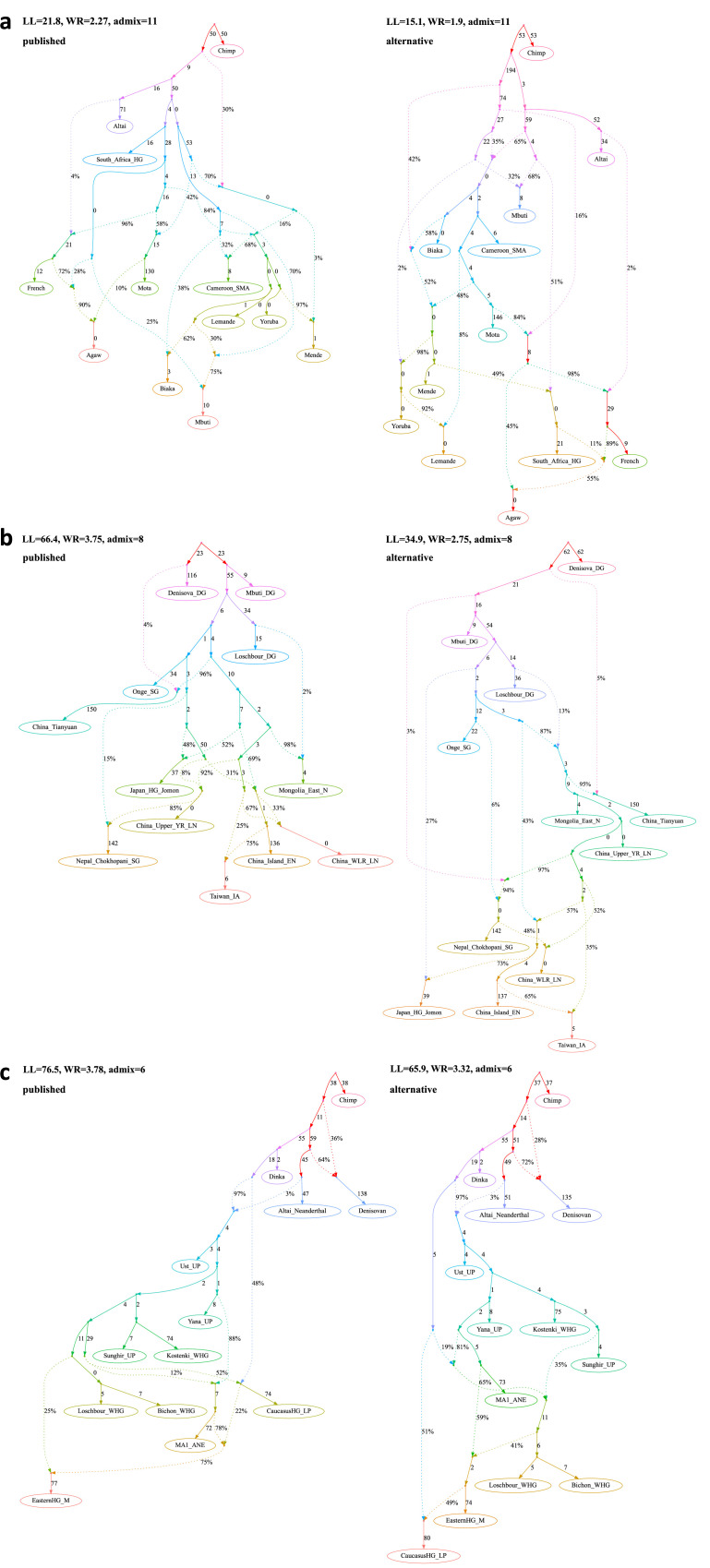
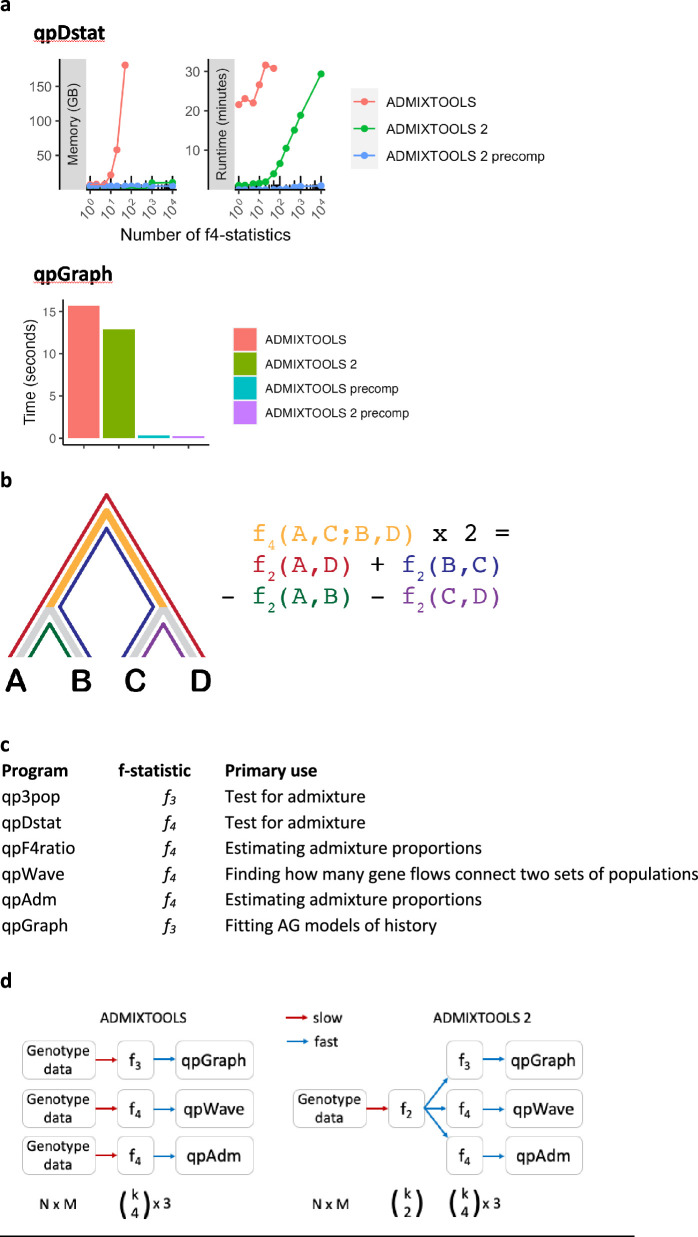
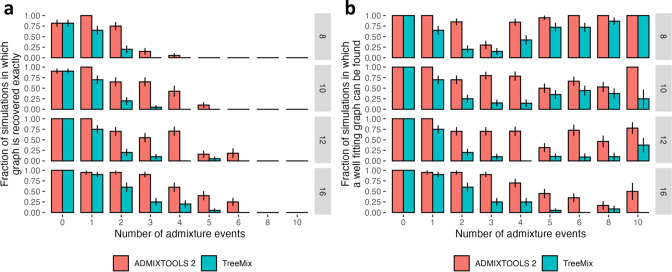
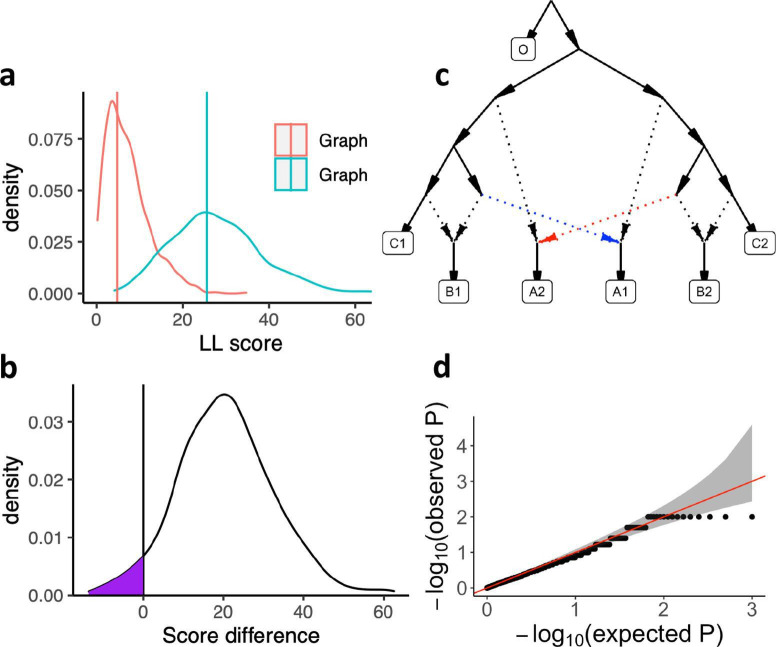
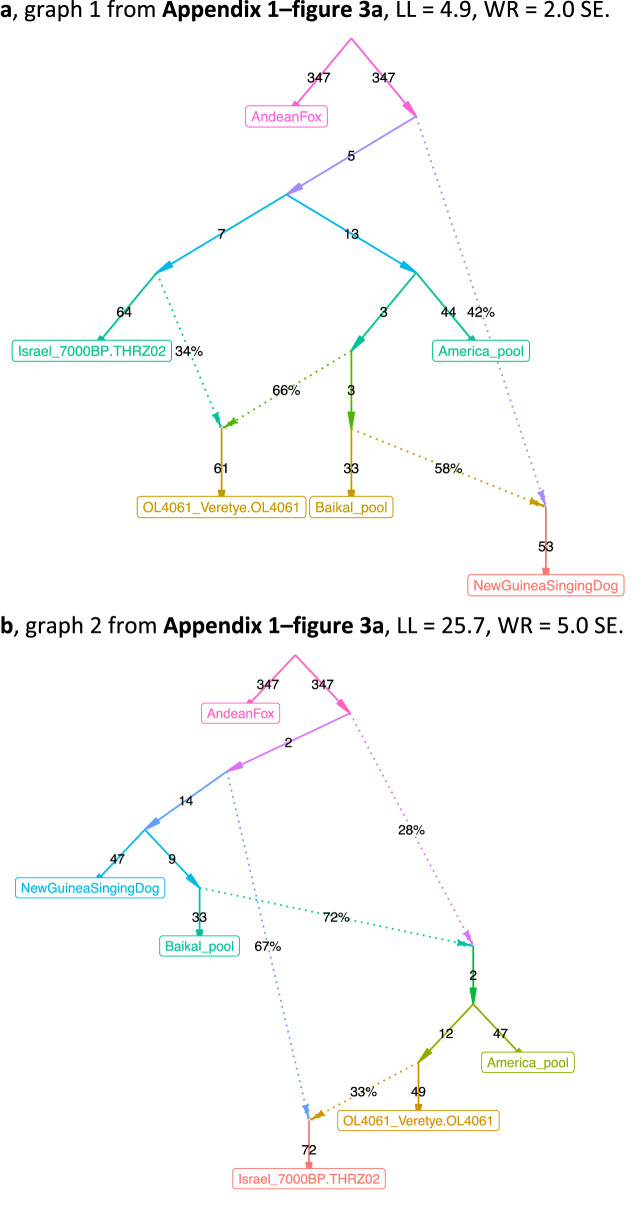
Our understanding of population history in deep time has been assisted by fitting admixture graphs (AGs) to data: models that specify the ordering of population splits and mixtures, which along with the amount of genetic drift and the proportions of mixture, is the only information needed to predict the patterns of allele frequency correlation among populations. The space of possible AGs relating populations is vast, and thus most published studies have identified fitting AGs through a manual process driven by prior hypotheses, leaving the majority of alternative models unexplored. Here, we develop a method for systematically searching the space of all AGs that can incorporate non-genetic information in the form of topology constraints. We implement this findGraphs tool within a software package, ADMIXTOOLS 2, which is a reimplementation of the ADMIXTOOLS software with new features and large performance gains. We apply this methodology to identify alternative models to AGs that played key roles in eight publications and find that in nearly all cases many alternative models fit nominally or significantly better than the published one. Our results suggest that strong claims about population history from AGs should only be made when all well-fitting and temporally plausible models share common topological features. Our re-evaluation of published data also provides insight into the population histories of humans, dogs, and horses, identifying features that are stable across the models we explored, as well as scenarios of populations relationships that differ in important ways from models that have been highlighted in the literature.
Keywords: admixture graphs; dogs; evolutionary biology; f-statistics; genetics; genomics; horses; human; humans; population genetics.
© 2023, Maier, Flegontov et al.
Conflict of interest statement
RM, PF, OF, UI, PC, DR No competing interests declared
Figures
Update of
References
-
- Baumdicker F, Bisschop G, Goldstein D, Gower G, Ragsdale AP, Tsambos G, Zhu S, Eldon B, Ellerman EC, Galloway JG, Gladstein AL, Gorjanc G, Guo B, Jeffery B, Kretzschumar WW, Lohse K, Matschiner M, Nelson D, Pope NS, Quinto-Cortés CD, Rodrigues MF, Saunack K, Sellinger T, Thornton K, van Kemenade H, Wohns AW, Wong Y, Gravel S, Kern AD, Koskela J, Ralph PL, Kelleher J. Efficient ancestry and mutation simulation with msprime 1.0. Genetics. 2022;220:iyab229. doi: 10.1093/genetics/iyab229. - DOI - PMC - PubMed
-
- Bellwood P. The checkered prehistory of rice movement southwards as a domesticated cereal—from the yangzi to the equator. Rice. 2011;4:93–103. doi: 10.1007/s12284-011-9068-9. - DOI
-
- Bergström A, Frantz L, Schmidt R, Ersmark E, Lebrasseur O, Girdland-Flink L, Lin AT, Storå J, Sjögren KG, Anthony D, Antipina E, Amiri S, Bar-Oz G, Bazaliiskii VI, Bulatović J, Brown D, Carmagnini A, Davy T, Fedorov S, Fiore I, Fulton D, Germonpré M, Haile J, Irving-Pease EK, Jamieson A, Janssens L, Kirillova I, Horwitz LK, Kuzmanovic-Cvetković J, Kuzmin Y, Losey RJ, Dizdar DL, Mashkour M, Novak M, Onar V, Orton D, Pasarić M, Radivojević M, Rajković D, Roberts B, Ryan H, Sablin M, Shidlovskiy F, Stojanović I, Tagliacozzo A, Trantalidou K, Ullén I, Villaluenga A, Wapnish P, Dobney K, Götherström A, Linderholm A, Dalén L, Pinhasi R, Larson G, Skoglund P. Origins and genetic legacy of prehistoric dogs. Science. 2020;370:557–564. doi: 10.1126/science.aba9572. - DOI - PMC - PubMed
-
- Bergström A, Stanton DWG, Taron UH, Frantz L, Sinding M-HS, Ersmark E, Pfrengle S, Cassatt-Johnstone M, Lebrasseur O, Girdland-Flink L, Fernandes DM, Ollivier M, Speidel L, Gopalakrishnan S, Westbury MV, Ramos-Madrigal J, Feuerborn TR, Reiter E, Gretzinger J, Münzel SC, Swali P, Conard NJ, Carøe C, Haile J, Linderholm A, Androsov S, Barnes I, Baumann C, Benecke N, Bocherens H, Brace S, Carden RF, Drucker DG, Fedorov S, Gasparik M, Germonpré M, Grigoriev S, Groves P, Hertwig ST, Ivanova VV, Janssens L, Jennings RP, Kasparov AK, Kirillova IV, Kurmaniyazov I, Kuzmin YV, Kosintsev PA, Lázničková-Galetová M, Leduc C, Nikolskiy P, Nussbaumer M, O’Drisceoil C, Orlando L, Outram A, Pavlova EY, Perri AR, Pilot M, Pitulko VV, Plotnikov VV, Protopopov AV, Rehazek A, Sablin M, Seguin-Orlando A, Storå J, Verjux C, Zaibert VF, Zazula G, Crombé P, Hansen AJ, Willerslev E, Leonard JA, Götherström A, Pinhasi R, Schuenemann VJ, Hofreiter M, Gilbert MTP, Shapiro B, Larson G, Krause J, Dalén L, Skoglund P. Grey wolf genomic history reveals a dual ancestry of dogs. Nature. 2022;607:313–320. doi: 10.1038/s41586-022-04824-9. - DOI - PMC - PubMed
-
- Boos DD. Introduction to the bootstrap world. Statistical Science. 2003;18:168–174. doi: 10.1214/ss/1063994971. - DOI
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
