

Regulation of Monocyte Activation by PPARα Through Interaction With the cGAS-STING Pathway
- PMID: 37058417
- PMCID: PMC10281240
- DOI: 10.2337/db22-0654
Regulation of Monocyte Activation by PPARα Through Interaction With the cGAS-STING Pathway
Abstract
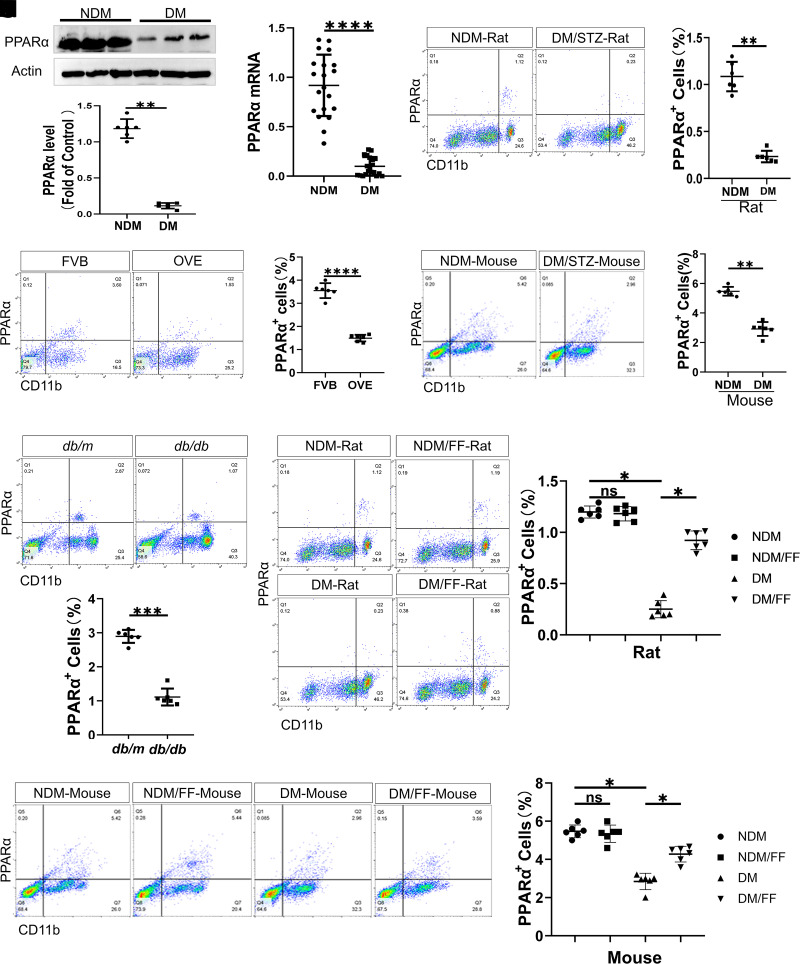
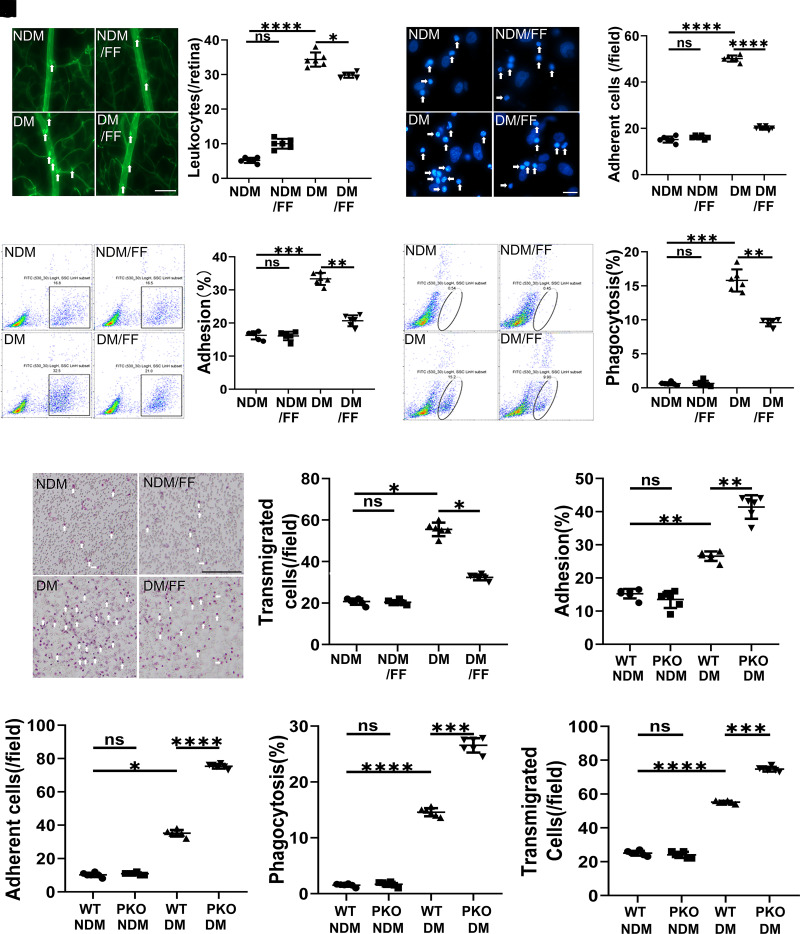
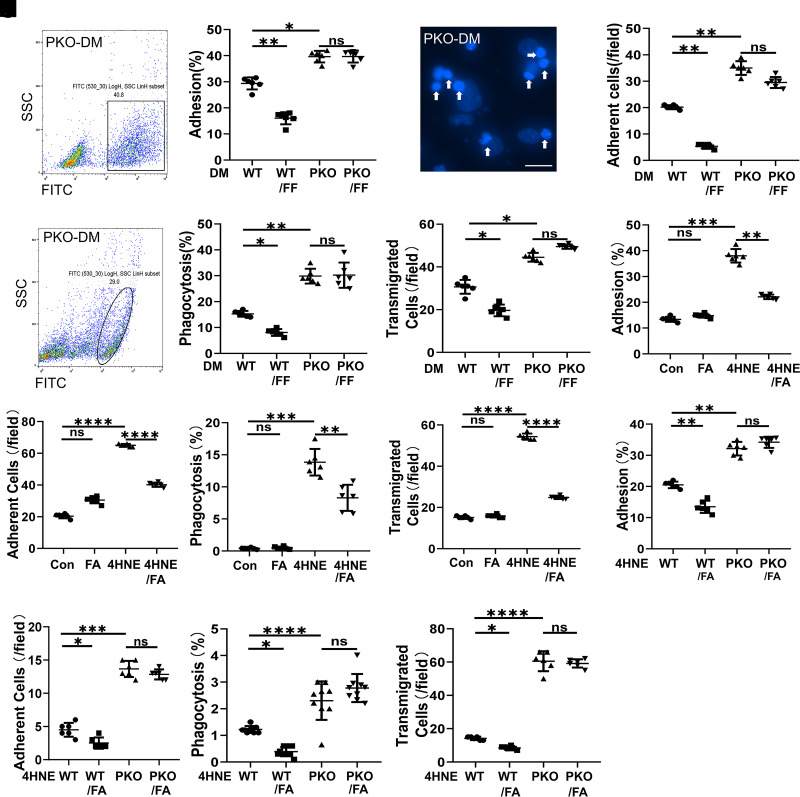
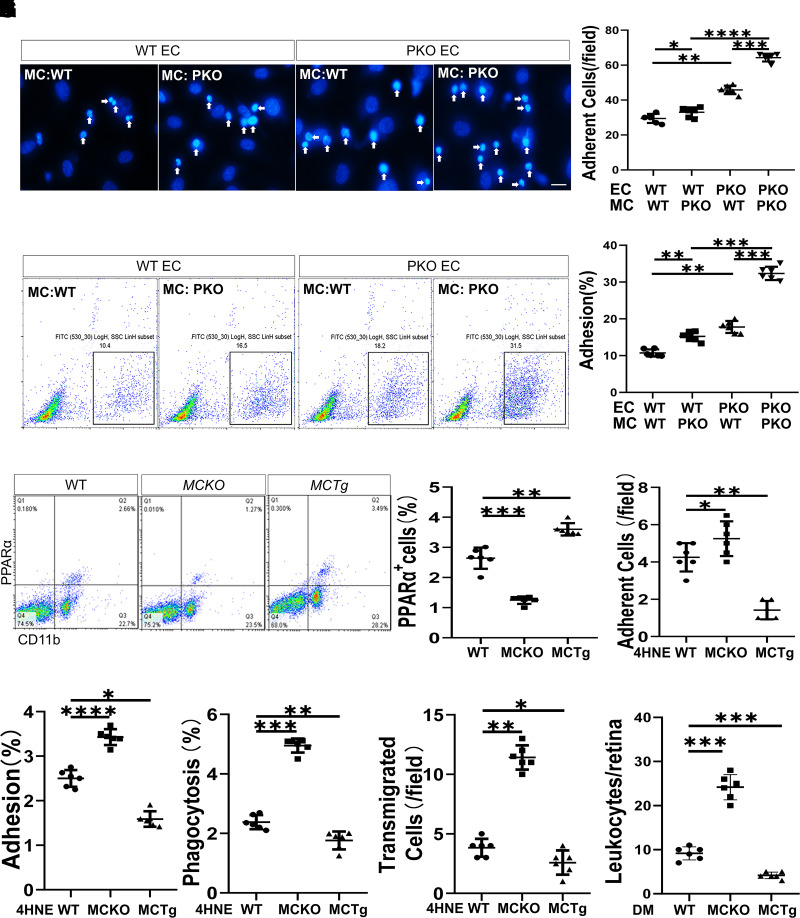
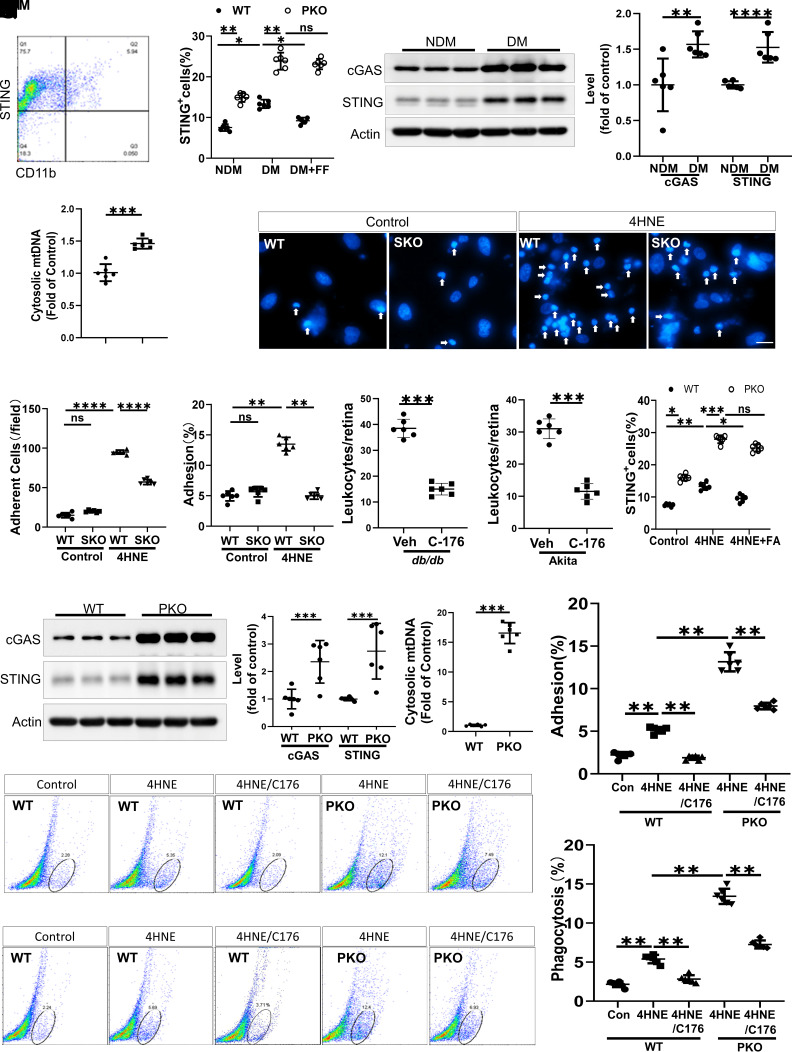
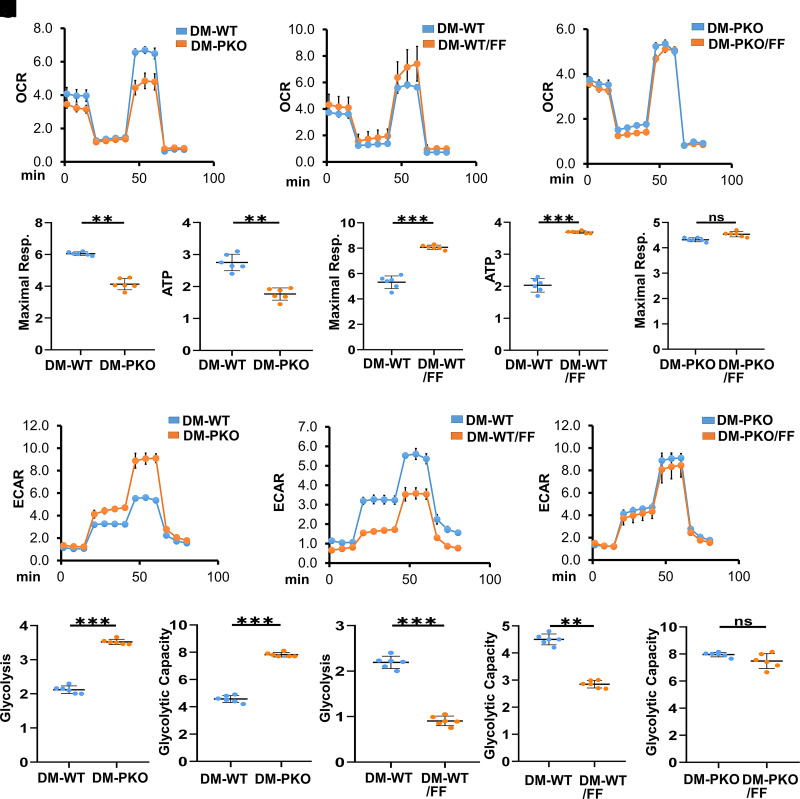
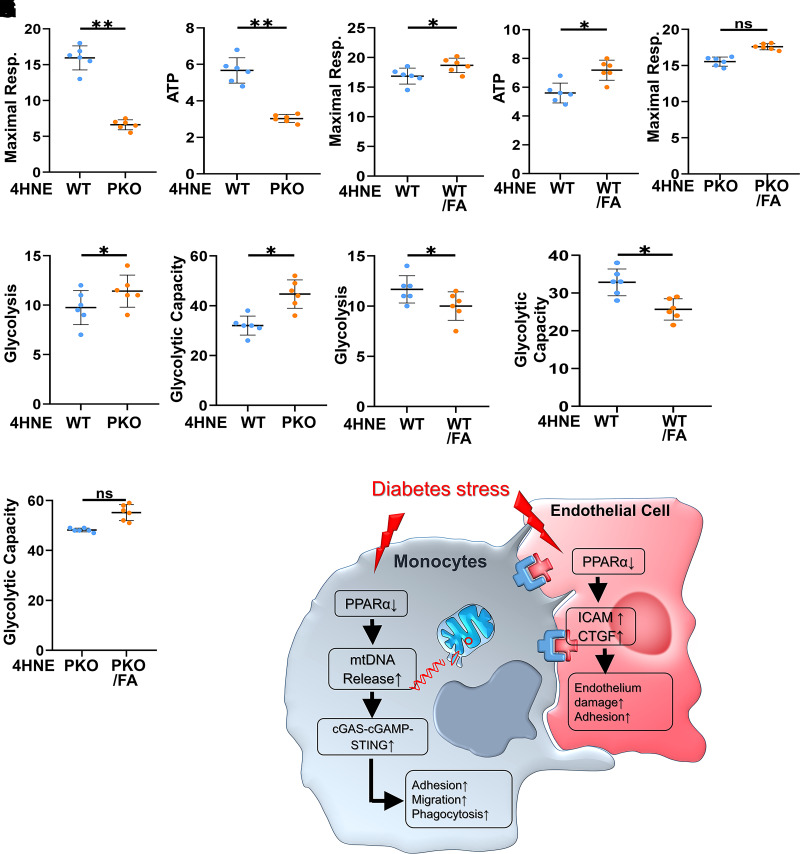
Monocyte activation plays an important role in diabetic complications such as diabetic retinopathy (DR). However, the regulation of monocyte activation in diabetes remains elusive. Fenofibrate, an agonist of peroxisome proliferator-activated receptor-α (PPARα), has shown robust therapeutic effects on DR in patients with type 2 diabetes. Here we found that PPARα levels were significantly downregulated in monocytes from patients with diabetes and animal models, correlating with monocyte activation. Fenofibrate attenuated monocyte activation in diabetes, while PPARα knockout alone induced monocyte activation. Furthermore, monocyte-specific PPARα overexpression ameliorated, while monocyte-specific PPARα knockout aggravated monocyte activation in diabetes. PPARα knockout impaired mitochondrial function while also increasing glycolysis in monocytes. PPARα knockout increased cytosolic mitochondrial DNA release and activation of the cyclic GMP-AMP synthase (cGAS)-stimulator of interferon genes (STING) pathway in monocytes under diabetic conditions. STING knockout or STING inhibitor attenuated monocyte activation induced by diabetes or by PPARα knockout. These observations suggest that PPARα negatively regulates monocyte activation through metabolic reprogramming and interaction with the cGAS-STING pathway.
© 2023 by the American Diabetes Association.
Conflict of interest statement
Figures
References
-
- Mesquida M, Drawnel F, Fauser S. The role of inflammation in diabetic eye disease. Semin Immunopathol 2019;41:427–445 - PubMed
-
- Binet F, Cagnone G, Crespo-Garcia S, et al. Neutrophil extracellular traps target senescent vasculature for tissue remodeling in retinopathy. Science 2020;369:eaay5356. - PubMed
-
- Rezzola S, Corsini M, Chiodelli P, et al. Inflammation and N-formyl peptide receptors mediate the angiogenic activity of human vitreous humour in proliferative diabetic retinopathy. Diabetologia 2017;60:719–728 - PubMed
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
