

CRISPR-Cas effector specificity and cleavage site determine phage escape outcomes
- PMID: 37058530
- PMCID: PMC10132644
- DOI: 10.1371/journal.pbio.3002065
CRISPR-Cas effector specificity and cleavage site determine phage escape outcomes
Abstract
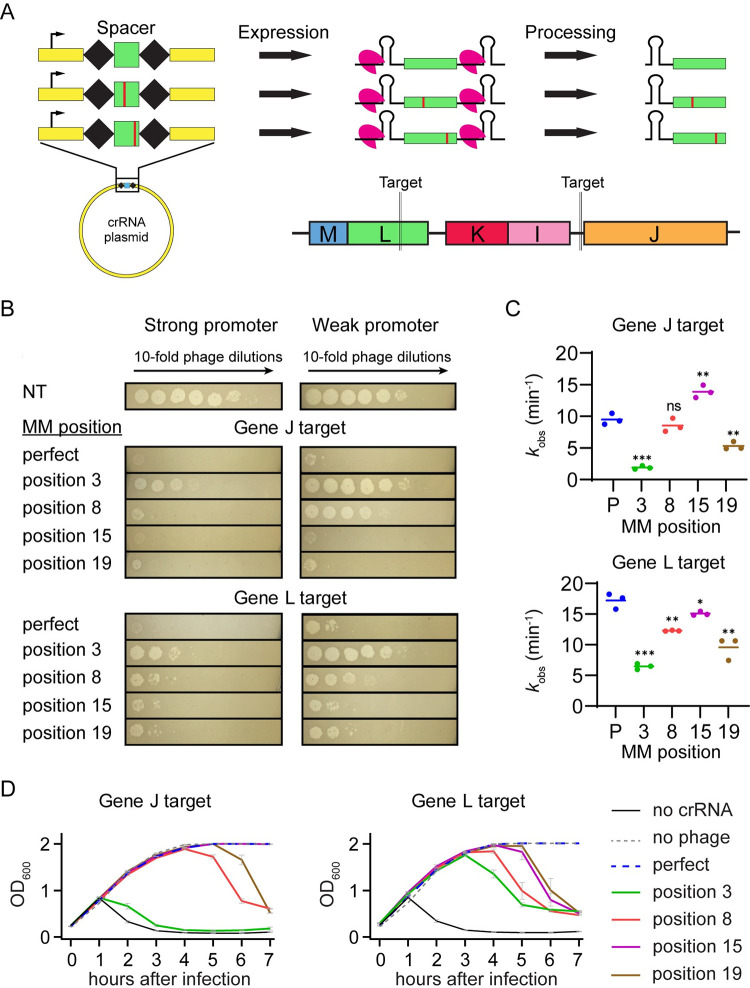
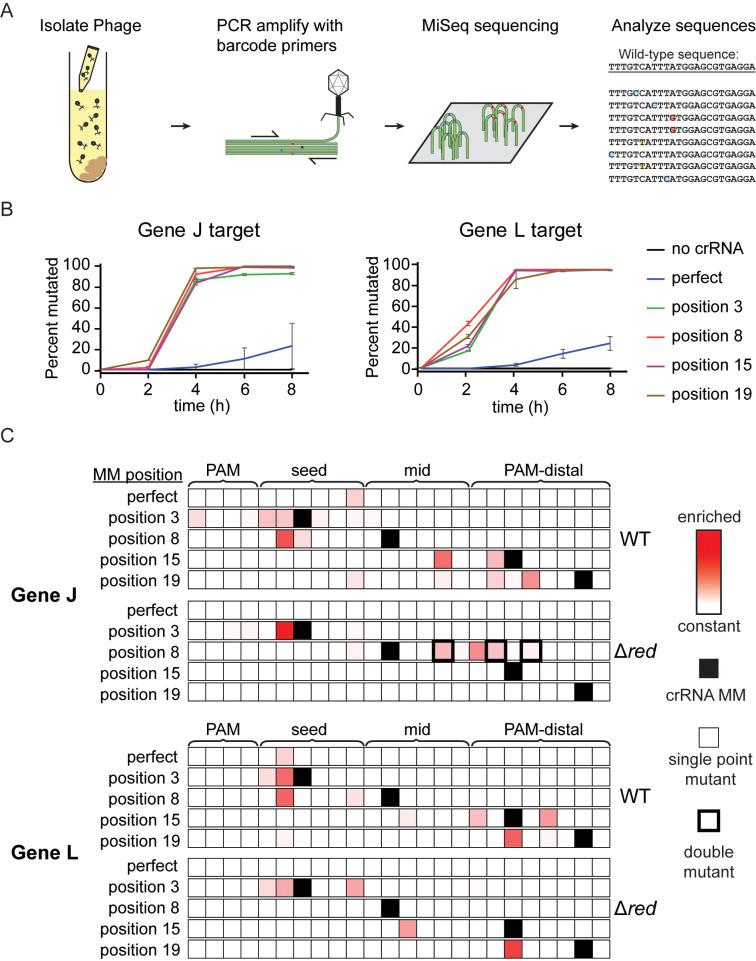
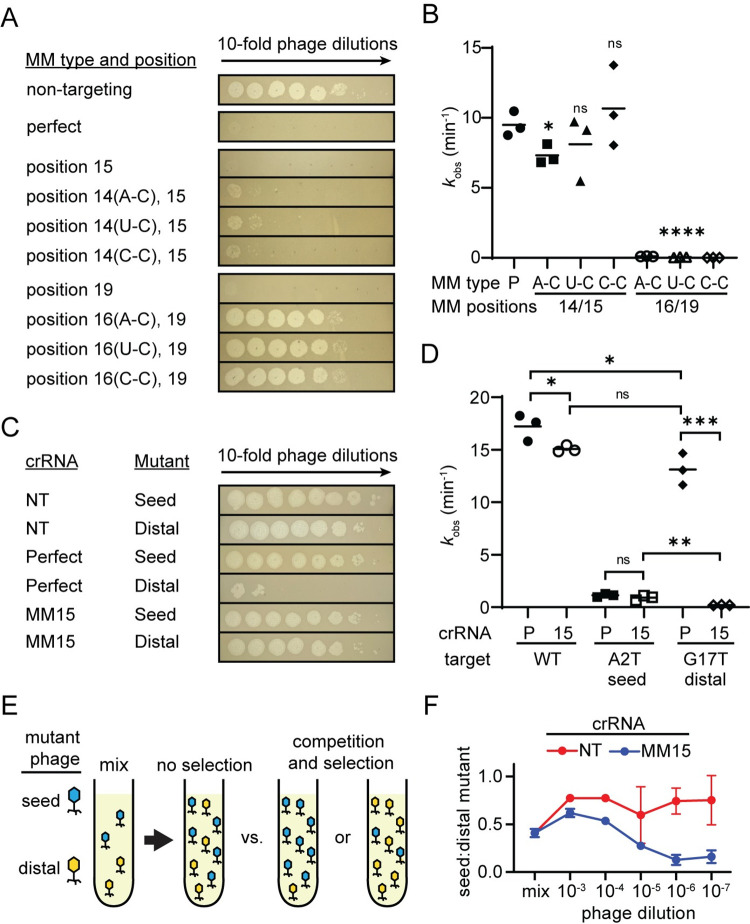
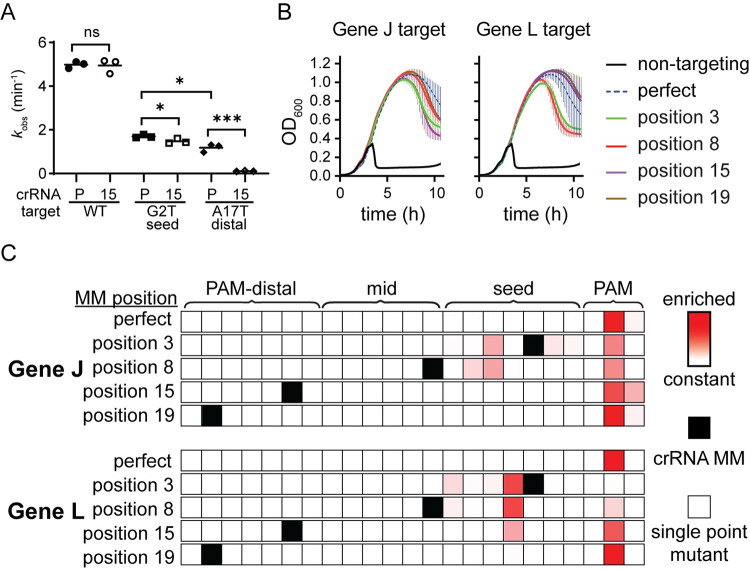
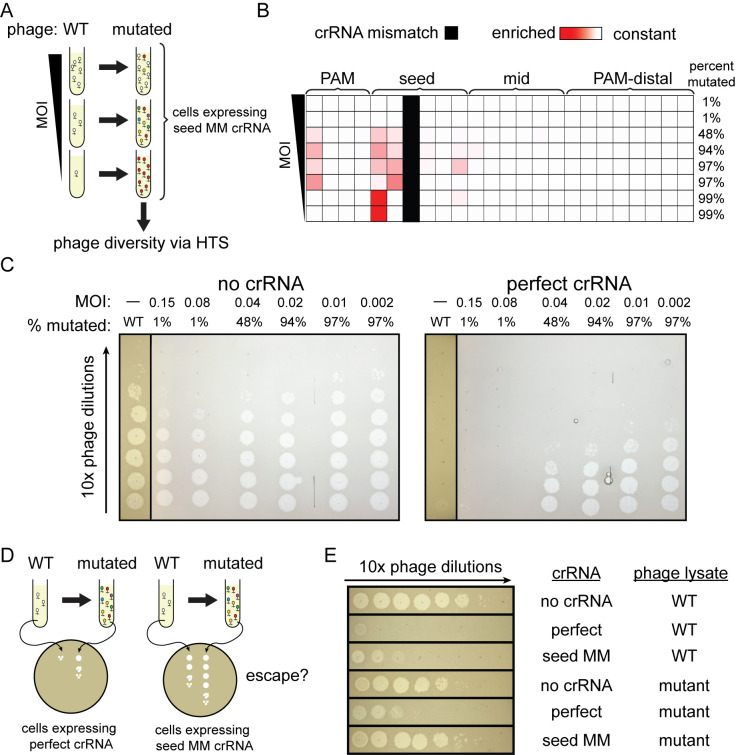
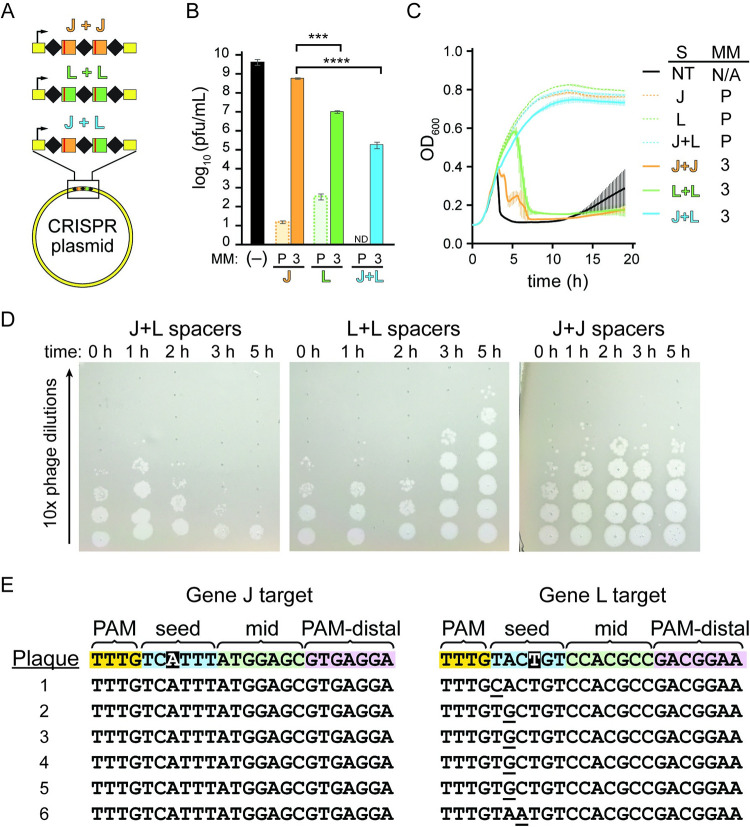
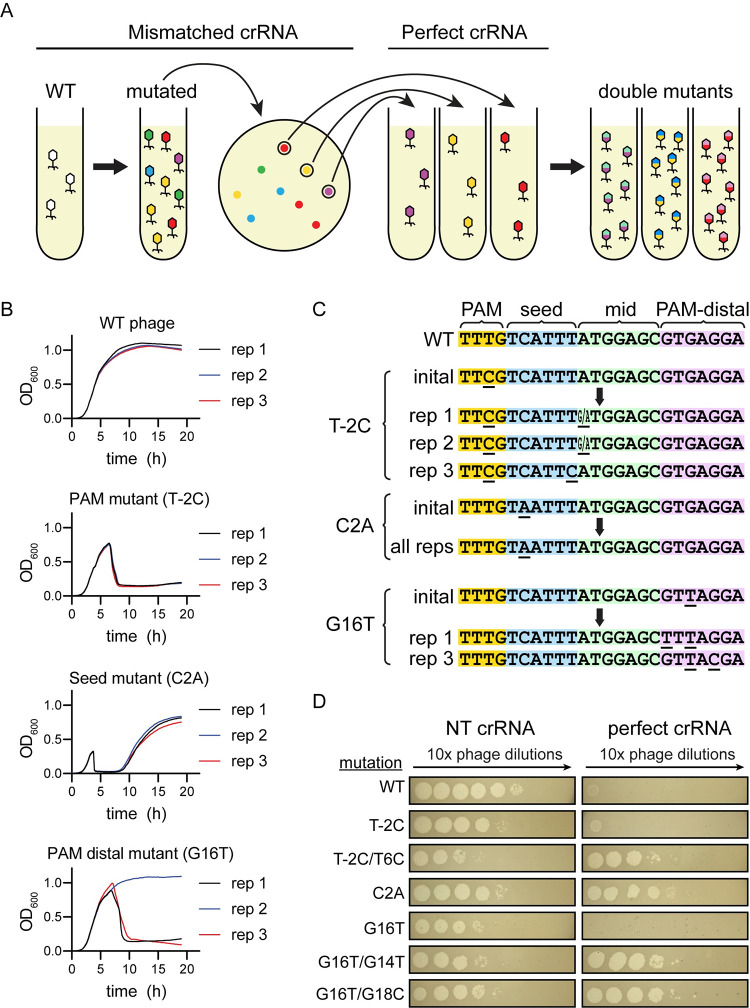
CRISPR-mediated interference relies on complementarity between a guiding CRISPR RNA (crRNA) and target nucleic acids to provide defense against bacteriophage. Phages escape CRISPR-based immunity mainly through mutations in the protospacer adjacent motif (PAM) and seed regions. However, previous specificity studies of Cas effectors, including the class 2 endonuclease Cas12a, have revealed a high degree of tolerance of single mismatches. The effect of this mismatch tolerance has not been extensively studied in the context of phage defense. Here, we tested defense against lambda phage provided by Cas12a-crRNAs containing preexisting mismatches against the genomic targets in phage DNA. We find that most preexisting crRNA mismatches lead to phage escape, regardless of whether the mismatches ablate Cas12a cleavage in vitro. We used high-throughput sequencing to examine the target regions of phage genomes following CRISPR challenge. Mismatches at all locations in the target accelerated emergence of mutant phage, including mismatches that greatly slowed cleavage in vitro. Unexpectedly, our results reveal that a preexisting mismatch in the PAM-distal region results in selection of mutations in the PAM-distal region of the target. In vitro cleavage and phage competition assays show that dual PAM-distal mismatches are significantly more deleterious than combinations of seed and PAM-distal mismatches, resulting in this selection. However, similar experiments with Cas9 did not result in emergence of PAM-distal mismatches, suggesting that cut-site location and subsequent DNA repair may influence the location of escape mutations within target regions. Expression of multiple mismatched crRNAs prevented new mutations from arising in multiple targeted locations, allowing Cas12a mismatch tolerance to provide stronger and longer-term protection. These results demonstrate that Cas effector mismatch tolerance, existing target mismatches, and cleavage site strongly influence phage evolution.
Copyright: © 2023 Schelling et al. This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
