

Mitochondrial membrane potential instability on reperfusion after ischemia does not depend on mitochondrial Ca2+ uptake
- PMID: 37061004
- PMCID: PMC10206190
- DOI: 10.1016/j.jbc.2023.104708
Mitochondrial membrane potential instability on reperfusion after ischemia does not depend on mitochondrial Ca2+ uptake
Abstract
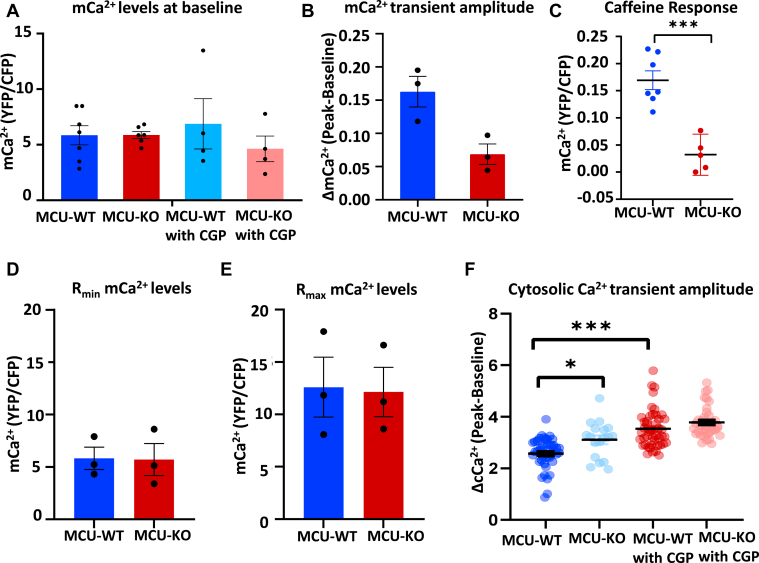
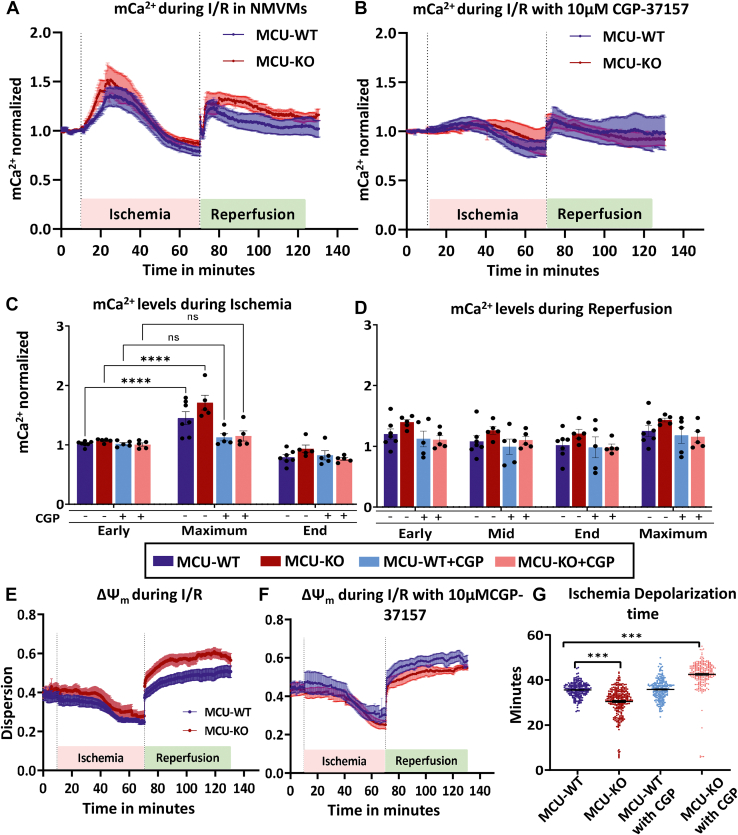
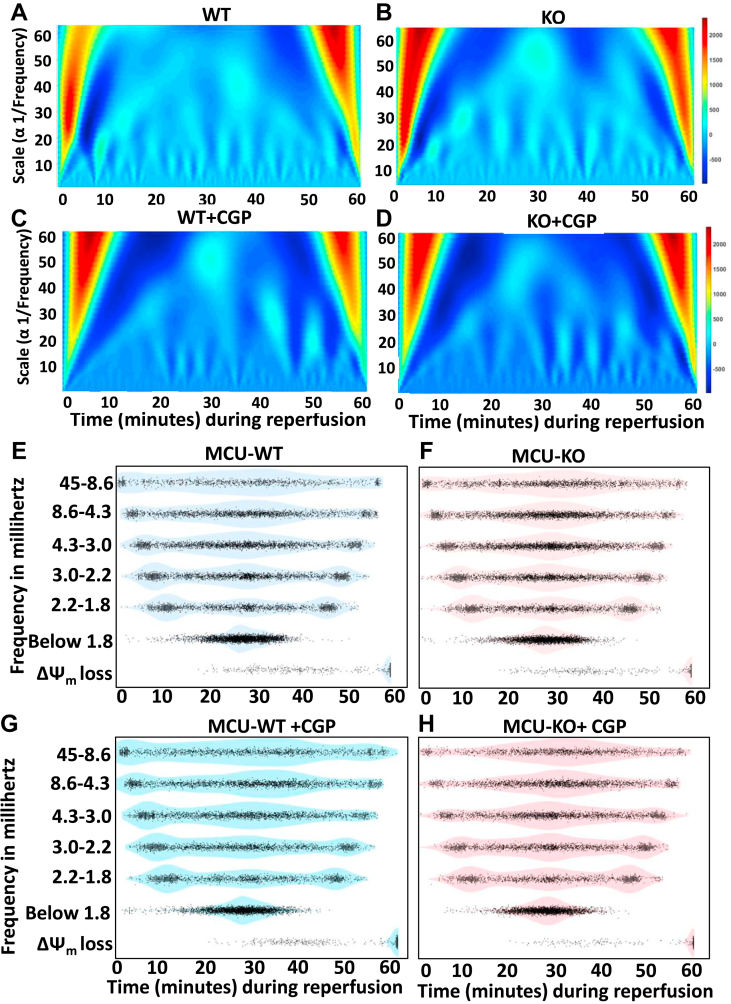
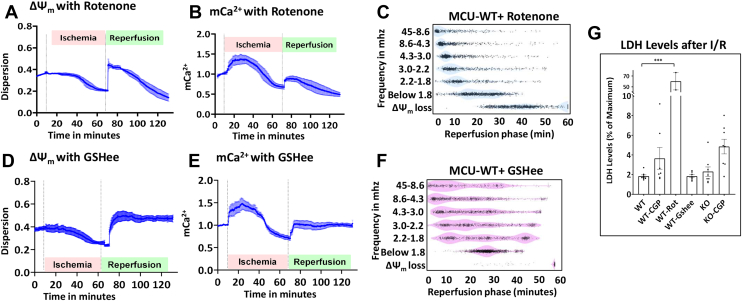
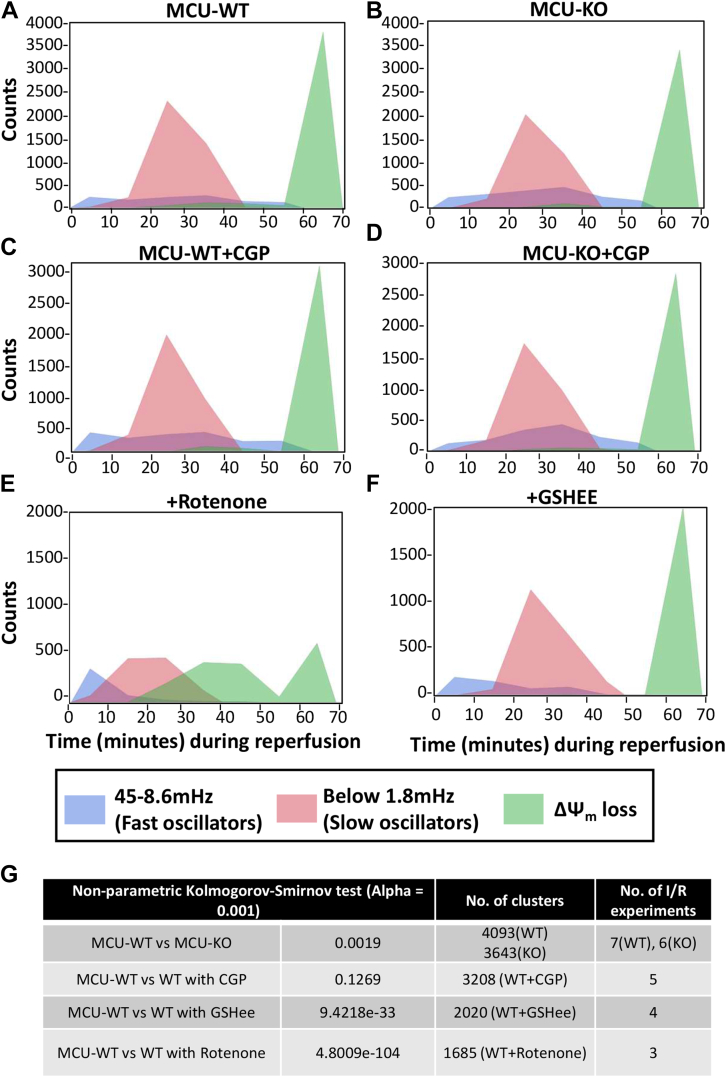
Physiologic Ca2+ entry via the Mitochondrial Calcium Uniporter (MCU) participates in energetic adaption to workload but may also contribute to cell death during ischemia/reperfusion (I/R) injury. The MCU has been identified as the primary mode of Ca2+ import into mitochondria. Several groups have tested the hypothesis that Ca2+ import via MCU is detrimental during I/R injury using genetically-engineered mouse models, yet the results from these studies are inconclusive. Furthermore, mitochondria exhibit unstable or oscillatory membrane potentials (ΔΨm) when subjected to stress, such as during I/R, but it is unclear if the primary trigger is an excess influx of mitochondrial Ca2+ (mCa2+), reactive oxygen species (ROS) accumulation, or other factors. Here, we critically examine whether MCU-mediated mitochondrial Ca2+ uptake during I/R is involved in ΔΨm instability, or sustained mitochondrial depolarization, during reperfusion by acutely knocking out MCU in neonatal mouse ventricular myocyte (NMVM) monolayers subjected to simulated I/R. Unexpectedly, we find that MCU knockout does not significantly alter mCa2+ import during I/R, nor does it affect ΔΨm recovery during reperfusion. In contrast, blocking the mitochondrial sodium-calcium exchanger (mNCE) suppressed the mCa2+ increase during Ischemia but did not affect ΔΨm recovery or the frequency of ΔΨm oscillations during reperfusion, indicating that mitochondrial ΔΨm instability on reperfusion is not triggered by mCa2+. Interestingly, inhibition of mitochondrial electron transport or supplementation with antioxidants stabilized I/R-induced ΔΨm oscillations. The findings are consistent with mCa2+ overload being mediated by reverse-mode mNCE activity and supporting ROS-induced ROS release as the primary trigger of ΔΨm instability during reperfusion injury.
Keywords: image processing; ischemia; mitochondrial membrane potential; oscillation; oxidative phosphorylation; reperfusion; time-series analysis; wavelet.
Copyright © 2023 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Conflict of interests The authors declare no conflict of interest with the contents of this article.
Figures

Similar articles
-
Inhibition of MCU forces extramitochondrial adaptations governing physiological and pathological stress responses in heart.Proc Natl Acad Sci U S A. 2015 Jul 21;112(29):9129-34. doi: 10.1073/pnas.1504705112. Epub 2015 Jul 7. Proc Natl Acad Sci U S A. 2015. PMID: 26153425 Free PMC article.
-
Distinct mPTP activation mechanisms in ischaemia-reperfusion: contributions of Ca2+, ROS, pH, and inorganic polyphosphate.Cardiovasc Res. 2015 May 1;106(2):237-48. doi: 10.1093/cvr/cvv097. Epub 2015 Mar 5. Cardiovasc Res. 2015. PMID: 25742913 Free PMC article.
-
Mitochondrial instability during regional ischemia-reperfusion underlies arrhythmias in monolayers of cardiomyocytes.J Mol Cell Cardiol. 2015 Jan;78:90-9. doi: 10.1016/j.yjmcc.2014.09.024. Epub 2014 Sep 28. J Mol Cell Cardiol. 2015. PMID: 25268650 Free PMC article.
-
Mitochondrial Bioenergetics During Ischemia and Reperfusion.Adv Exp Med Biol. 2017;982:141-167. doi: 10.1007/978-3-319-55330-6_8. Adv Exp Med Biol. 2017. PMID: 28551786 Review.
-
Mitochondrial calcium and the regulation of metabolism in the heart.J Mol Cell Cardiol. 2015 Jan;78:35-45. doi: 10.1016/j.yjmcc.2014.10.019. Epub 2014 Nov 7. J Mol Cell Cardiol. 2015. PMID: 25450609 Free PMC article. Review.
Cited by
-
Monitoring mitochondrial calcium in cardiomyocytes during coverslip hypoxia using a fluorescent lifetime indicator.J Mol Cell Cardiol Plus. 2024 Jun;8:100074. doi: 10.1016/j.jmccpl.2024.100074. Epub 2024 Apr 7. J Mol Cell Cardiol Plus. 2024. PMID: 38854449 Free PMC article.
-
Increased mitochondrial free Ca2+ during ischemia is suppressed, but not eliminated by, germline deletion of the mitochondrial Ca2+ uniporter.Cell Rep. 2023 Jul 25;42(7):112735. doi: 10.1016/j.celrep.2023.112735. Epub 2023 Jul 7. Cell Rep. 2023. PMID: 37421627 Free PMC article.
-
Health position paper and redox perspectives on reactive oxygen species as signals and targets of cardioprotection.Redox Biol. 2023 Nov;67:102894. doi: 10.1016/j.redox.2023.102894. Epub 2023 Oct 6. Redox Biol. 2023. PMID: 37839355 Free PMC article. Review.
-
How does mitochondrial Ca2+ change during ischemia and reperfusion? Implications for activation of the permeability transition pore.J Gen Physiol. 2025 Jan 6;157(1):e202313520. doi: 10.1085/jgp.202313520. Epub 2024 Dec 19. J Gen Physiol. 2025. PMID: 39699565 Review.
-
Mitochondrial calcium in cardiac ischemia/reperfusion injury and cardioprotection.Basic Res Cardiol. 2024 Aug;119(4):569-585. doi: 10.1007/s00395-024-01060-2. Epub 2024 Jun 19. Basic Res Cardiol. 2024. PMID: 38890208 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
