

Does AlphaFold2 model proteins' intracellular conformations? An experimental test using cross-linking mass spectrometry of endogenous ciliary proteins
- PMID: 37061613
- PMCID: PMC10105775
- DOI: 10.1038/s42003-023-04773-7
Does AlphaFold2 model proteins' intracellular conformations? An experimental test using cross-linking mass spectrometry of endogenous ciliary proteins
Abstract
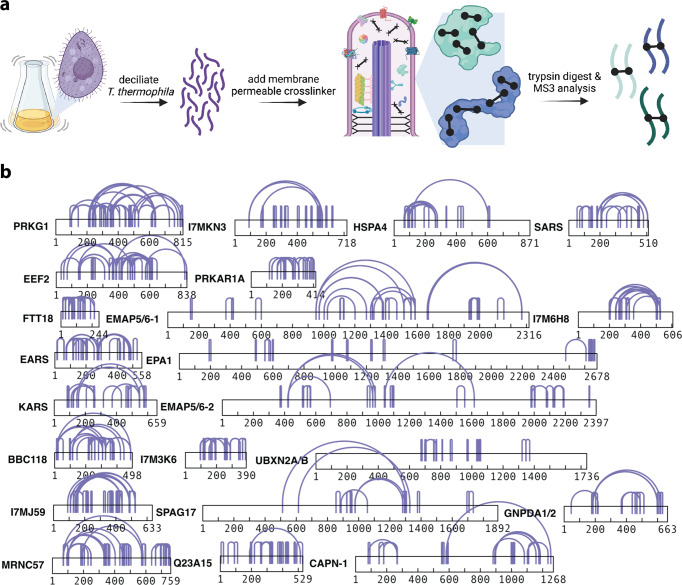
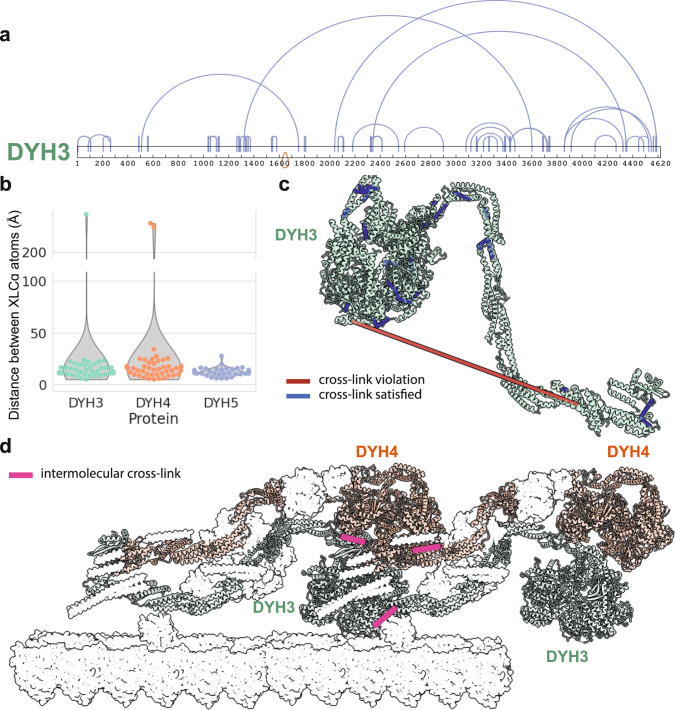
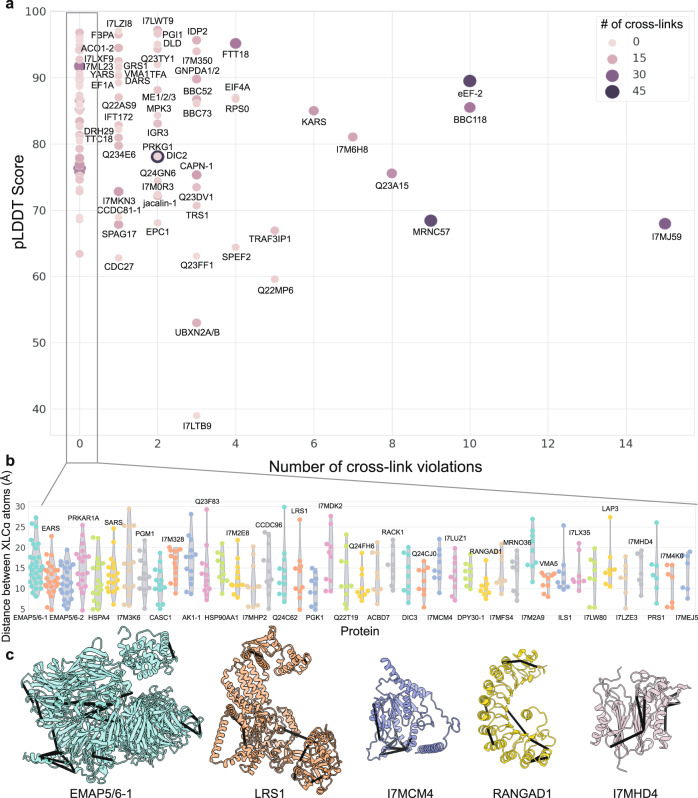
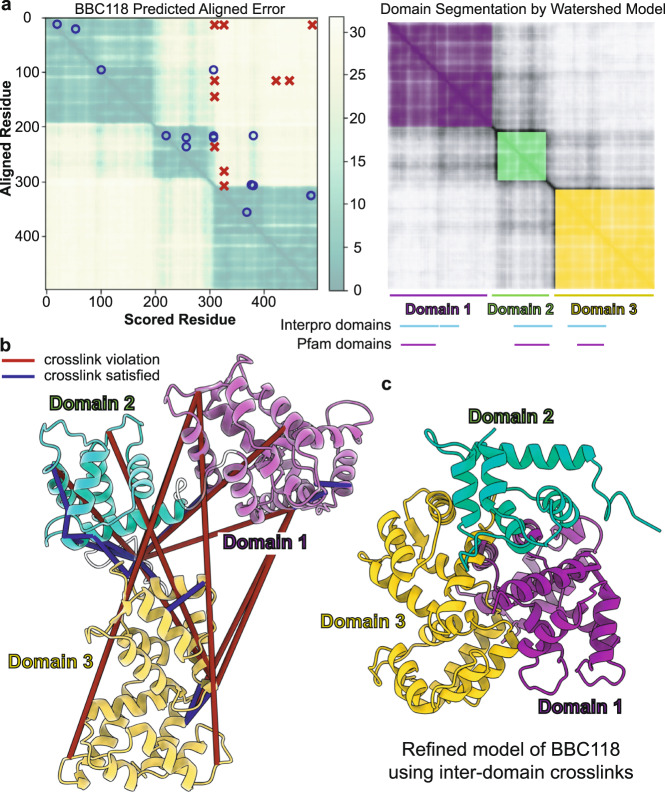
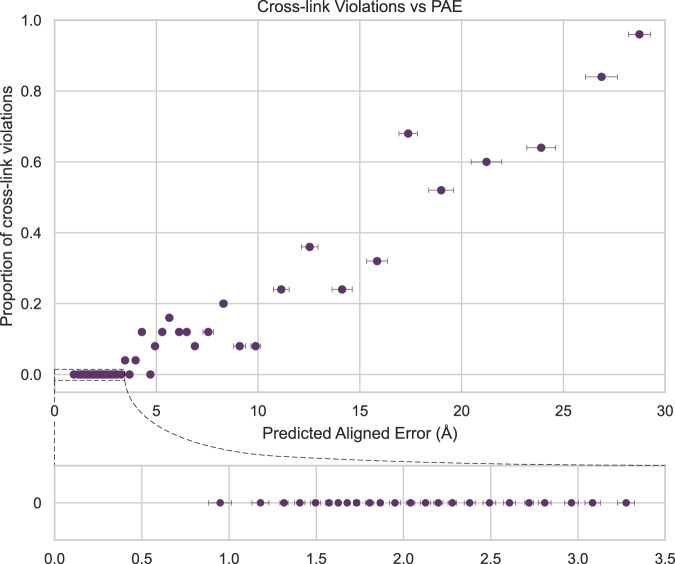
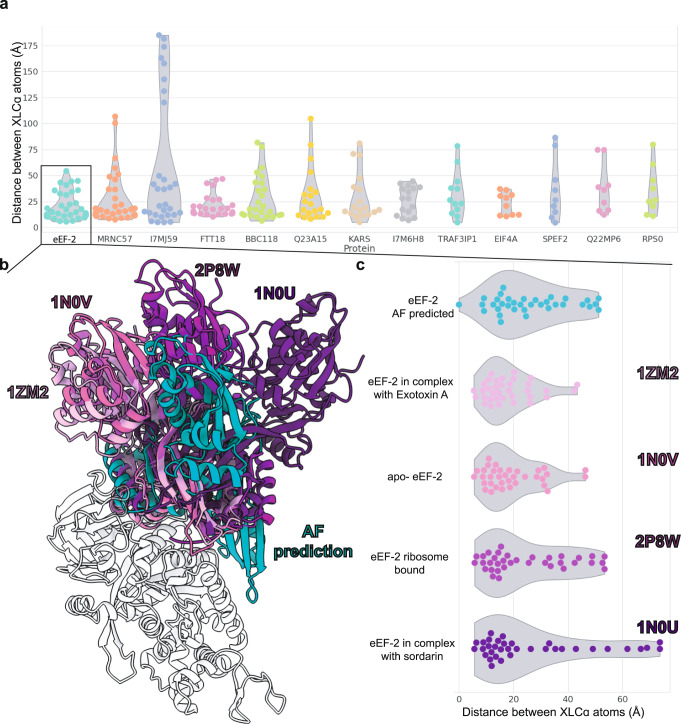
A major goal in structural biology is to understand protein assemblies in their biologically relevant states. Here, we investigate whether AlphaFold2 structure predictions match native protein conformations. We chemically cross-linked proteins in situ within intact Tetrahymena thermophila cilia and native ciliary extracts, identifying 1,225 intramolecular cross-links within the 100 best-sampled proteins, providing a benchmark of distance restraints obeyed by proteins in their native assemblies. The corresponding structure predictions were highly concordant, positioning 86.2% of cross-linked residues within Cɑ-to-Cɑ distances of 30 Å, consistent with the cross-linker length. 43% of proteins showed no violations. Most inconsistencies occurred in low-confidence regions or between domains. Overall, AlphaFold2 predictions with lower predicted aligned error corresponded to more correct native structures. However, we observe cases where rigid body domains are oriented incorrectly, as for ciliary protein BBC118, suggesting that combining structure prediction with experimental information will better reveal biologically relevant conformations.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
In Situ Structural Restraints from Cross-Linking Mass Spectrometry in Human Mitochondria.J Proteome Res. 2020 Jan 3;19(1):327-336. doi: 10.1021/acs.jproteome.9b00541. Epub 2019 Dec 19. J Proteome Res. 2020. PMID: 31746214 Free PMC article.
-
Small angle X-ray scattering and cross-linking for data assisted protein structure prediction in CASP 12 with prospects for improved accuracy.Proteins. 2018 Mar;86 Suppl 1(Suppl 1):202-214. doi: 10.1002/prot.25452. Epub 2018 Feb 7. Proteins. 2018. PMID: 29314274 Free PMC article.
-
The Conformational Preference of Chemical Cross-linkers Determines the Cross-linking Probability of Reactive Protein Residues.J Phys Chem B. 2020 Jun 4;124(22):4446-4453. doi: 10.1021/acs.jpcb.0c02522. Epub 2020 May 19. J Phys Chem B. 2020. PMID: 32369371
-
Chemical cross-linking and native mass spectrometry: A fruitful combination for structural biology.Protein Sci. 2015 Aug;24(8):1193-209. doi: 10.1002/pro.2696. Epub 2015 May 27. Protein Sci. 2015. PMID: 25970732 Free PMC article. Review.
-
Leveraging crosslinking mass spectrometry in structural and cell biology.Structure. 2022 Jan 6;30(1):37-54. doi: 10.1016/j.str.2021.11.007. Epub 2021 Dec 10. Structure. 2022. PMID: 34895473 Review.
Cited by
-
Functional implication of the homotrimeric multidomain vacuolar sorting receptor 1 (VSR1) from Arabidopsis thaliana.Sci Rep. 2024 Apr 26;14(1):9622. doi: 10.1038/s41598-024-57975-2. Sci Rep. 2024. PMID: 38671060 Free PMC article.
-
A ternary complex of MIPs in the A-tubule of basal bodies and axonemes depends on RIB22 and the EF-hand domain of RIB72A in Tetrahymena cilia.Mol Biol Cell. 2025 Apr 1;36(4):br13. doi: 10.1091/mbc.E24-12-0557. Epub 2025 Feb 12. Mol Biol Cell. 2025. PMID: 39937672 Free PMC article.
-
Mass spectrometry-complemented molecular modeling predicts the interaction interface for a camelid single-domain antibody targeting the Plasmodium falciparum circumsporozoite protein's C-terminal domain.Comput Struct Biotechnol J. 2024 Aug 28;23:3300-3314. doi: 10.1016/j.csbj.2024.08.023. eCollection 2024 Dec. Comput Struct Biotechnol J. 2024. PMID: 39296809 Free PMC article.
-
AlphaFold two years on: Validation and impact.Proc Natl Acad Sci U S A. 2024 Aug 20;121(34):e2315002121. doi: 10.1073/pnas.2315002121. Epub 2024 Aug 12. Proc Natl Acad Sci U S A. 2024. PMID: 39133843 Free PMC article.
-
A new discrete-geometry approach for integrative docking of proteins using chemical crosslinks.bioRxiv [Preprint]. 2025 Jan 7:2024.10.24.619977. doi: 10.1101/2024.10.24.619977. bioRxiv. 2025. Update in: J Chem Inf Model. 2025 May 12;65(9):4576-4592. doi: 10.1021/acs.jcim.4c02412. PMID: 39553940 Free PMC article. Updated. Preprint.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
