

Reduction of TRPC1/TRPC3 mediated Ca2+-signaling protects oxidative stress-induced COPD
- PMID: 37062436
- PMCID: PMC10542863
- DOI: 10.1016/j.cellsig.2023.110681
Reduction of TRPC1/TRPC3 mediated Ca2+-signaling protects oxidative stress-induced COPD
Abstract
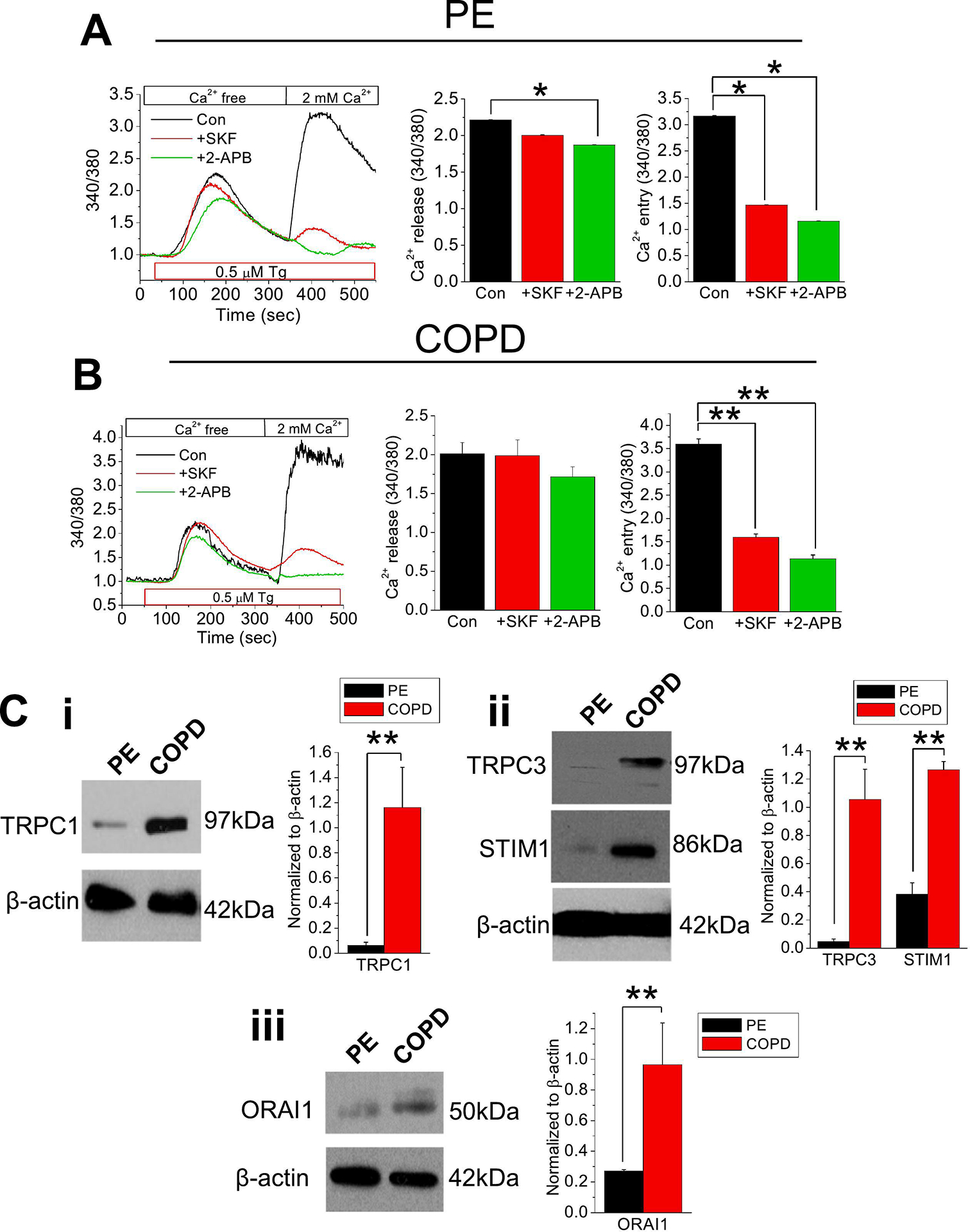
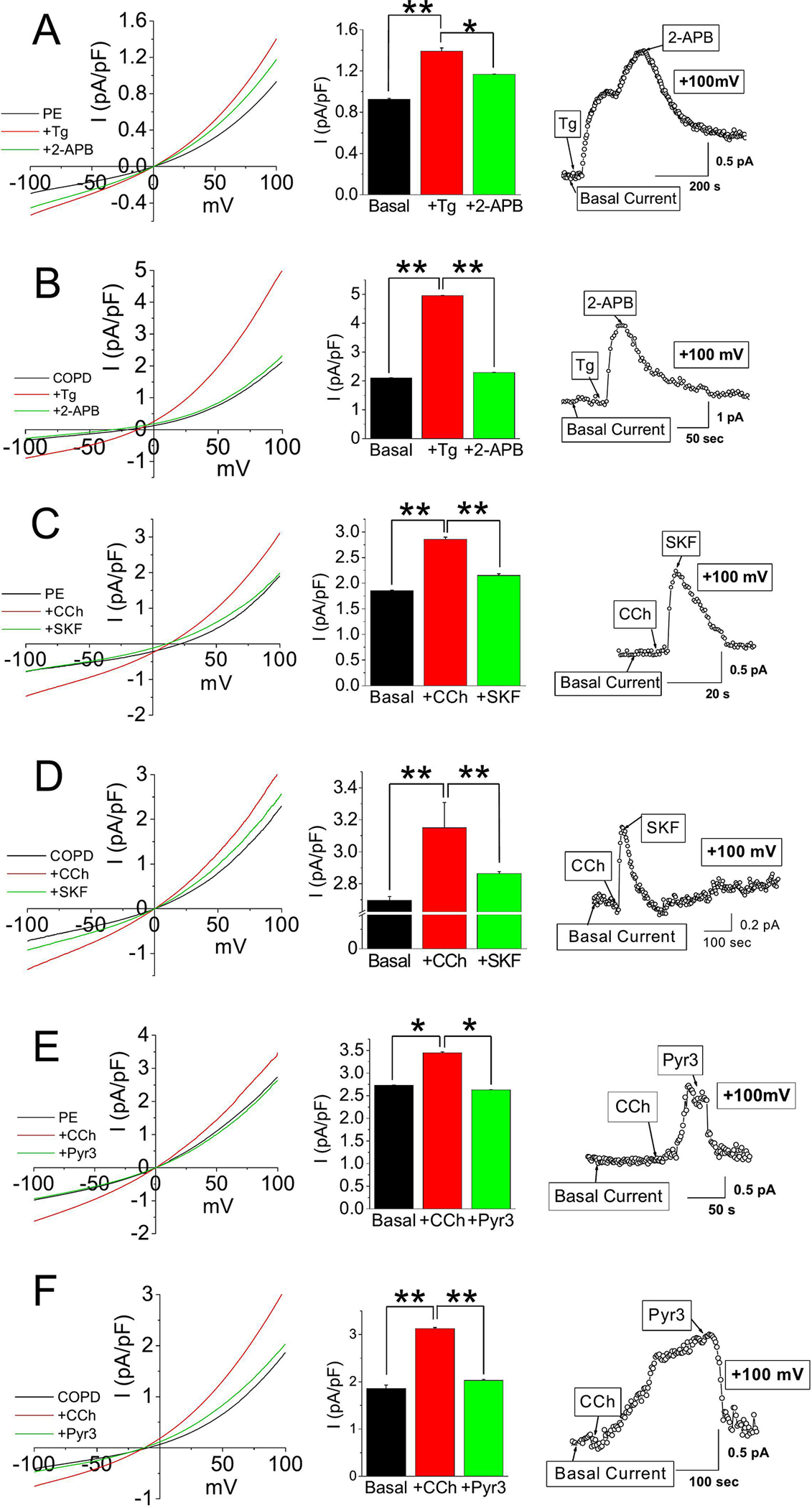
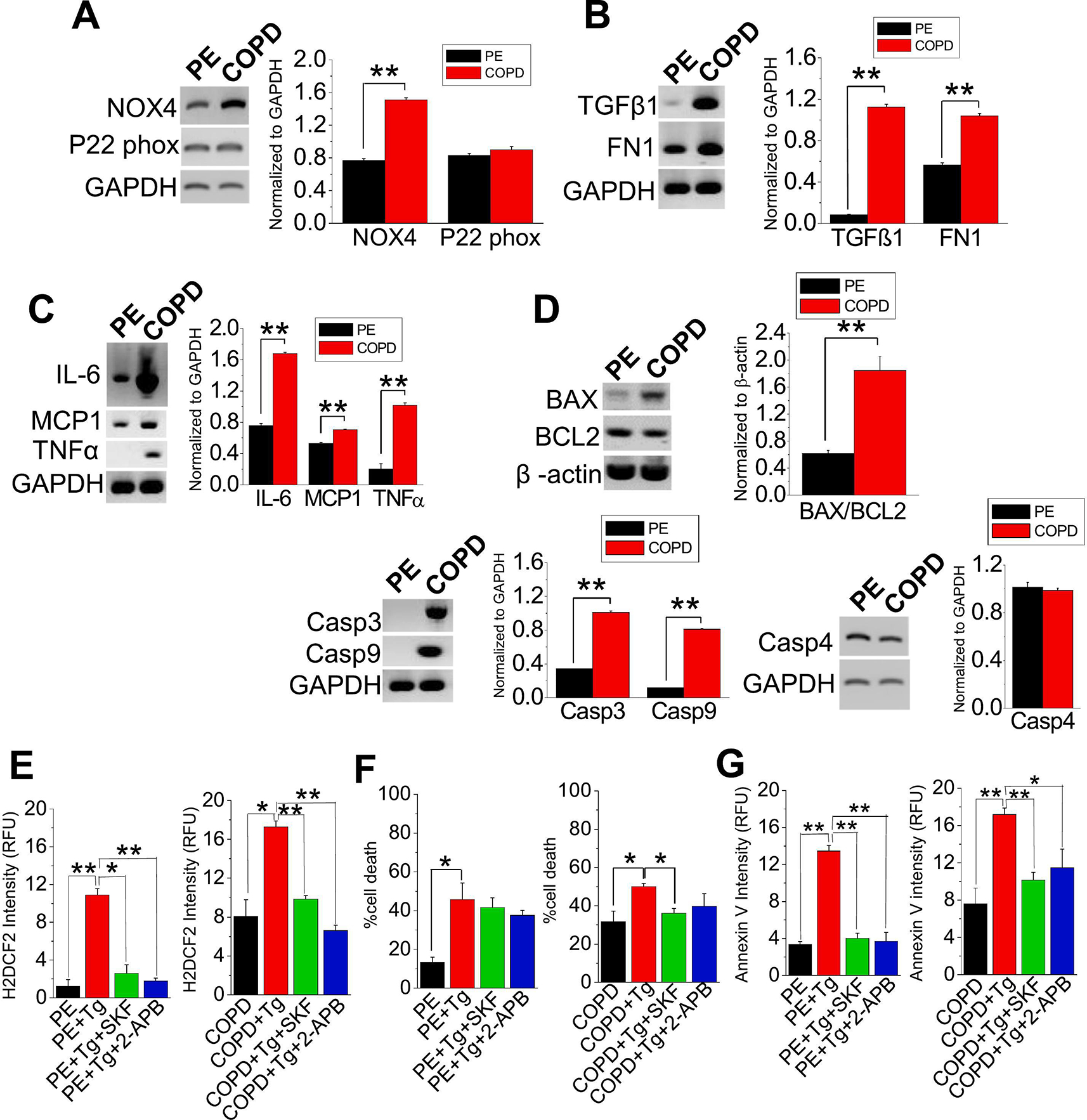
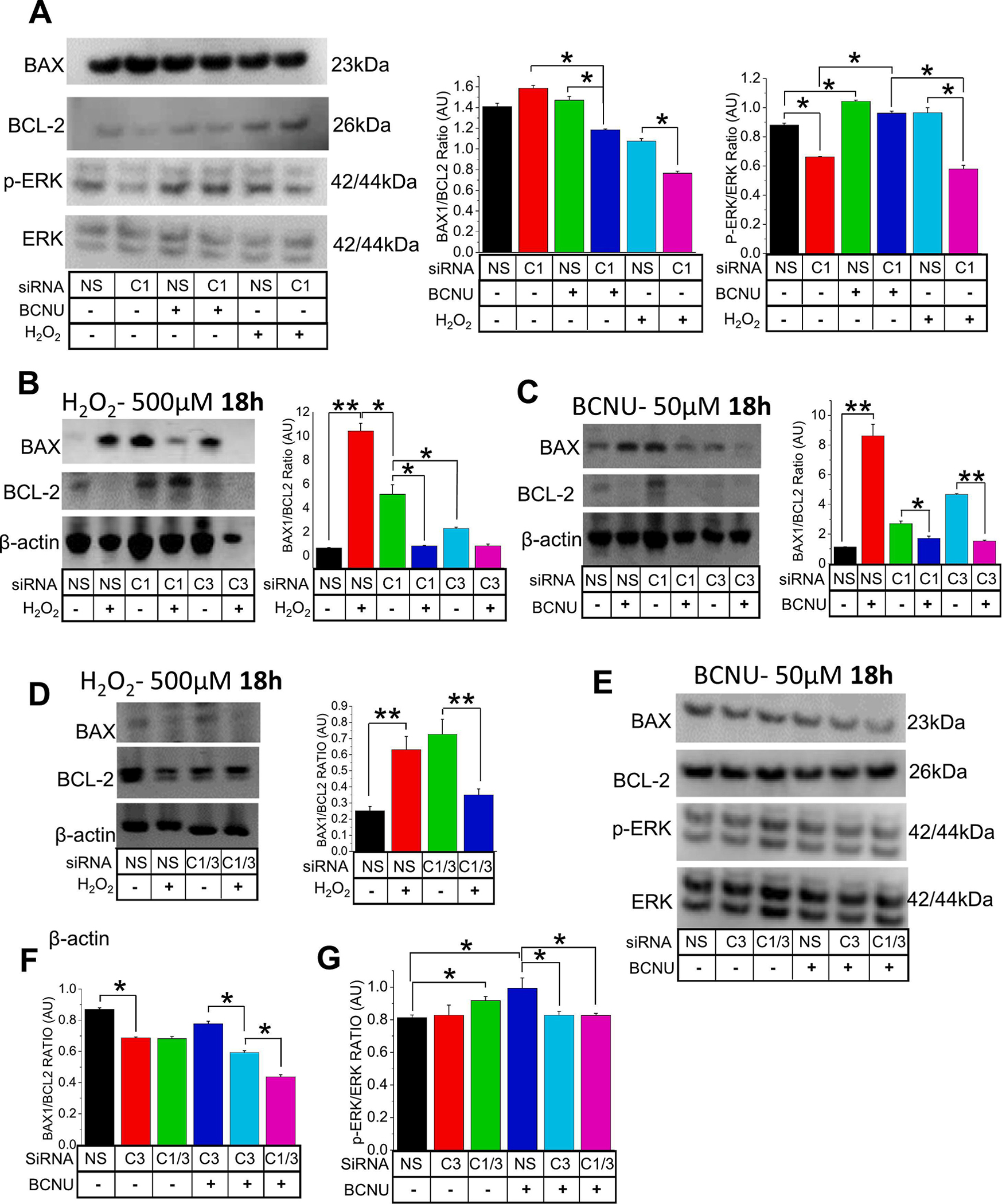
Oxidative stress is a predisposing factor in Chronic Obstructive Pulmonary Disease (COPD). Specifically, pulmonary epithelial (PE) cells reduce antioxidant capacity during COPD because of the continuous production of reactive oxygen species (ROS). However, the molecular pathogenesis that governs such ROS activity is unclear. Here we show that the dysregulation of intracellular calcium concentration ([Ca2+]i) in PE cells from COPD patients, compared to the healthy PE cells, is associated with the robust functional expressions of Transient Receptor Potential Canonical (TRPC)1 and TRPC3 channels, and Ca2+ entry (SOCE) components, Stromal Interaction Molecule 1 (STIM1) and ORAI1 channels. Additionally, the elevated expression levels of fibrotic, inflammatory, oxidative, and apoptotic markers in cells from COPD patients suggest detrimental pathway activation, thereby reducing the ability of lung remodeling. To further delineate the mechanism, we used human lung epithelial cell line, A549, since the behavior of SOCE and the expression patterns of TRPC1/C3, STIM1, and ORAI1 were much like PE cells. Notably, the knockdown of TRPC1/C3 in A549 cells substantially reduced the SOCE-induced [Ca2+]i rise, and reversed the ROS-mediated oxidative, fibrotic, inflammatory, and apoptotic responses, thus confirming the role of TRPC1/C3 in SOCE driven COPD-like condition. Higher TRPC1/C3, STIM1, and ORAI1 expressions, along with a greater Ca2+ entry, via SOCE in ROS-induced A549 cells, led to the rise in oxidative, fibrotic, inflammatory, and apoptotic gene expression, specifically through the extracellular signal-regulated kinase (ERK) pathway. Abatement of TRPC1 and/or TRPC3 reduced the mobilization of [Ca2+]i and reversed apoptotic gene expression and ERK activation, signifying the involvement of TRPC1/C3. Together these data suggest that TRPC1/C3 and SOCE facilitate the COPD condition through ROS-mediated cell death, thus implicating their likely roles as potential therapeutic targets for COPD. SUMMARY: Alterations in Ca2+ signaling modalities in normal pulmonary epithelial cells exhibit COPD through oxidative stress and cellular injury, compromising repair, which was alleviated through inhibition of store-operated calcium entry. SUBJECT AREA: Calcium, ROS, Cellular signaling, lung disease.
Keywords: Ca(2+) signaling; Lung disease; Oxidative stress; Pulmonary epithelial cells; TRPC channel.
Published by Elsevier Inc.
Conflict of interest statement
Declaration of Competing Interest The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Figures




References
-
- Brown DM, Donaldson K, Borm PJ, Schins RP, Dehnhardt M, Gilmour P, Jimenez LA, Stone V, Calcium and ROS-mediated activation of transcription factors and TNF-alpha cytokine gene expression in macrophages exposed to ultrafine particles, Am. J. Physiol. Lung Cell. Mol. Physiol. 2004; 286: L344–353. 10.1152/ajplung.00139.2003. - DOI - PubMed
-
- Cha M-J, Jang J-K, Ham O, Song B-W, Lee S-Y, Lee CY, Park J-H, Lee J, Seo H-H, Choi E, Jeon W, Hwang HJ, Shin H-T, Choi E, Hwang K-C, MicroRNA-145 suppresses ROS-induced Ca2+ overload of cardiomyocytes by targeting CaMKIIδ, Biochem. Biophys. Res. Commun. 2013; 435: 720–726. 10.1016/j.bbrc.2013.05.050. - DOI - PubMed
-
- Umemoto S, Tanaka M, Kawahara S, Kubo M, Umeji K, Hashimoto R, Matsuzaki M, Calcium antagonist reduces oxidative stress by upregulating Cu/Zn superoxide dismutase in stroke-prone spontaneously hypertensive rats, Hypertens. Res. Off. J. Jpn. Soc. Hypertens. 2004; 27: 877–885. 10.1291/hypres.27.877. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
