

MutaGAN: A sequence-to-sequence GAN framework to predict mutations of evolving protein populations
- PMID: 37066021
- PMCID: PMC10104372
- DOI: 10.1093/ve/vead022
MutaGAN: A sequence-to-sequence GAN framework to predict mutations of evolving protein populations
Abstract
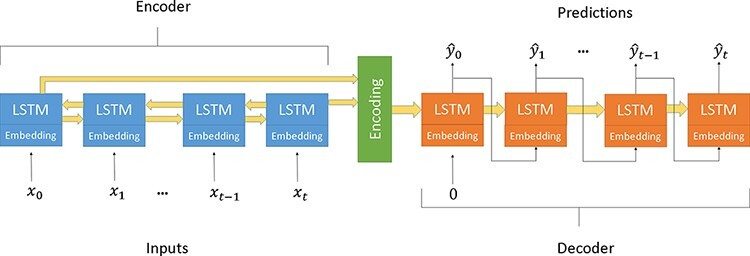
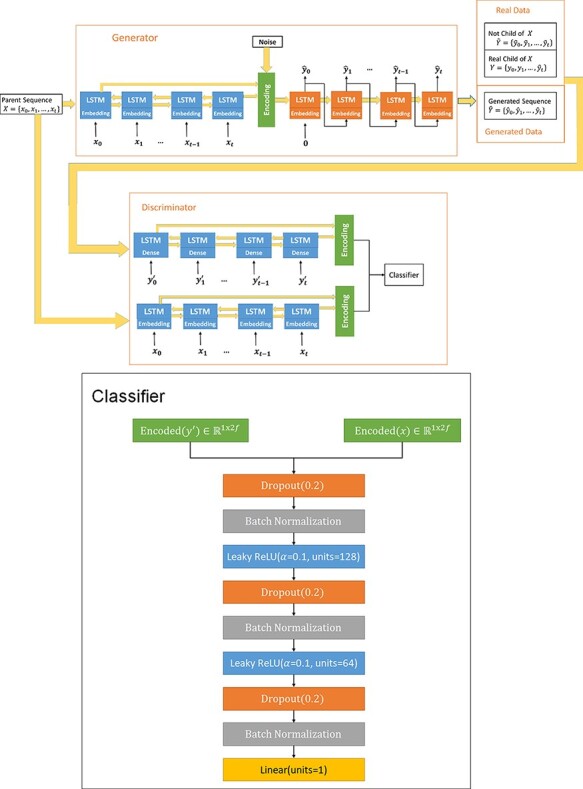
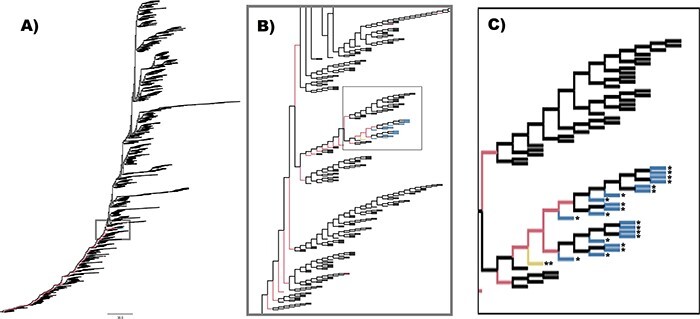
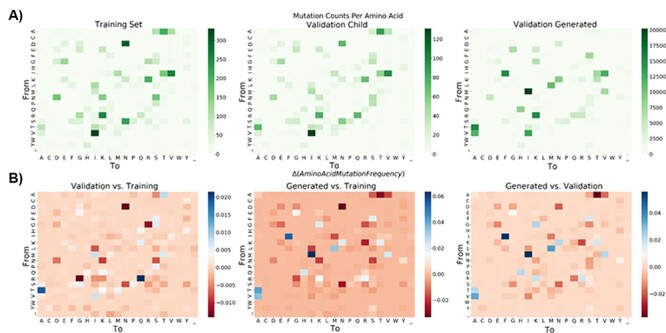
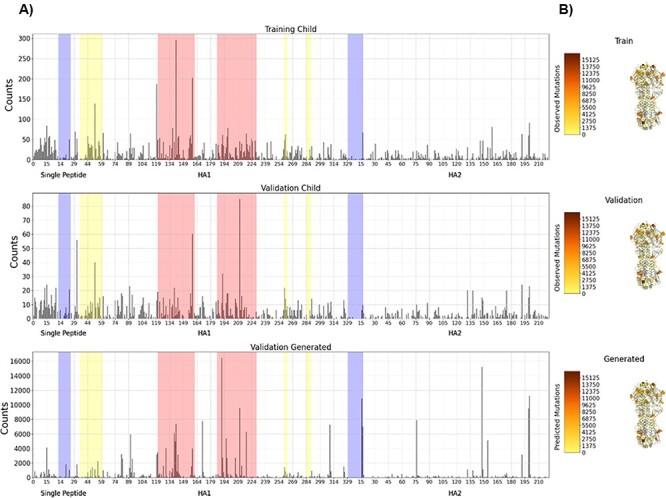
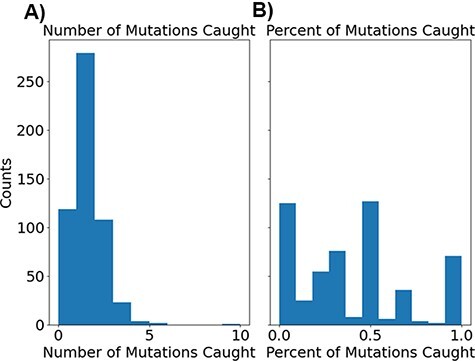
The ability to predict the evolution of a pathogen would significantly improve the ability to control, prevent, and treat disease. Machine learning, however, is yet to be used to predict the evolutionary progeny of a virus. To address this gap, we developed a novel machine learning framework, named MutaGAN, using generative adversarial networks with sequence-to-sequence, recurrent neural networks generator to accurately predict genetic mutations and evolution of future biological populations. MutaGAN was trained using a generalized time-reversible phylogenetic model of protein evolution with maximum likelihood tree estimation. MutaGAN was applied to influenza virus sequences because influenza evolves quickly and there is a large amount of publicly available data from the National Center for Biotechnology Information's Influenza Virus Resource. MutaGAN generated 'child' sequences from a given 'parent' protein sequence with a median Levenshtein distance of 4.00 amino acids. Additionally, the generator was able to generate sequences that contained at least one known mutation identified within the global influenza virus population for 72.8 per cent of parent sequences. These results demonstrate the power of the MutaGAN framework to aid in pathogen forecasting with implications for broad utility in evolutionary prediction for any protein population.
Keywords: Influenza virus; deep learning; evolution; generative adversarial networks; sequence generation.
© The Author(s) 2023. Published by Oxford University Press.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
References
-
- Abadi M. et al. (2016) ‘Tensorflow: A system for large-scale machine learning’, in 12th {USENIX} Symposium on Operating Systems Design and Implementation ({OSDI} 16), pp. 265–83.
-
- Alipanahi B. et al. (2015) ‘Predicting the Sequence Specificities of DNA- and RNA-Binding Proteins by Deep Learning’, Nature Biotechnology, 33: 831–8. - PubMed
-
- Anand N., and Huang P. (2018) ‘Generative Modeling for Protein Structures’, Advances in Neural Information Processing Systems, 31: 7504–15.
-
- Arjovsky M., Chintala S., and Bottou L. (2017) ‘Wasserstein Generative Adversarial Networks’, in International Conference on Machine Learning, pp. 214–23.
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
