

This is a preprint.
Improved detection of aberrant splicing using the Intron Jaccard Index
- PMID: 37066374
- PMCID: PMC10104204
- DOI: 10.1101/2023.03.31.23287997
Improved detection of aberrant splicing using the Intron Jaccard Index
Update in
-
Improved detection of aberrant splicing with FRASER 2.0 and the intron Jaccard index.Am J Hum Genet. 2023 Dec 7;110(12):2056-2067. doi: 10.1016/j.ajhg.2023.10.014. Epub 2023 Nov 24. Am J Hum Genet. 2023. PMID: 38006880 Free PMC article.
Abstract
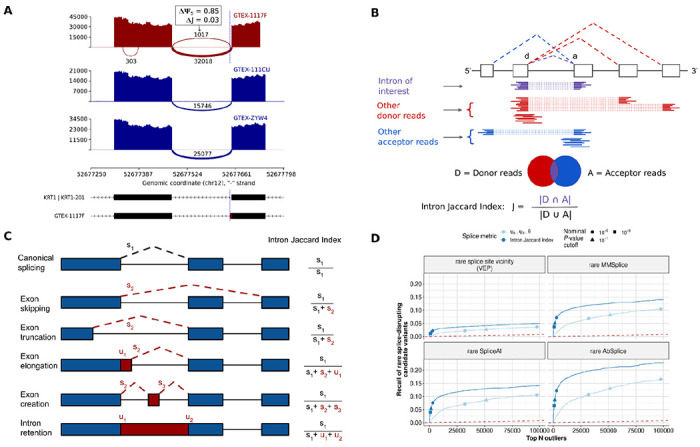
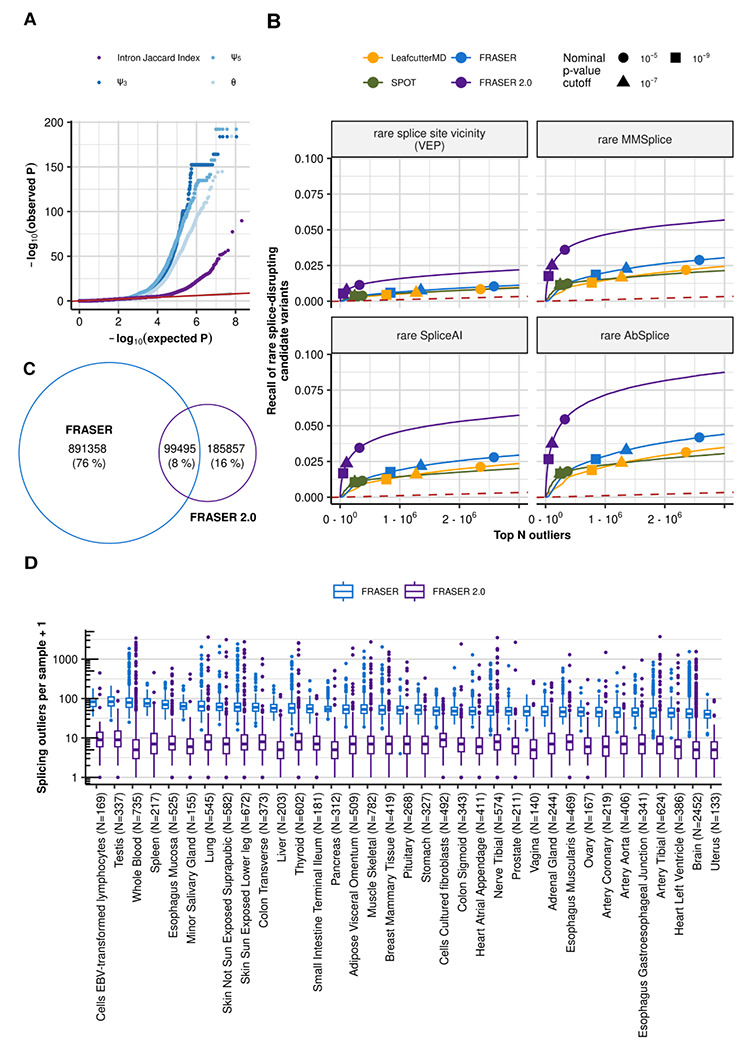
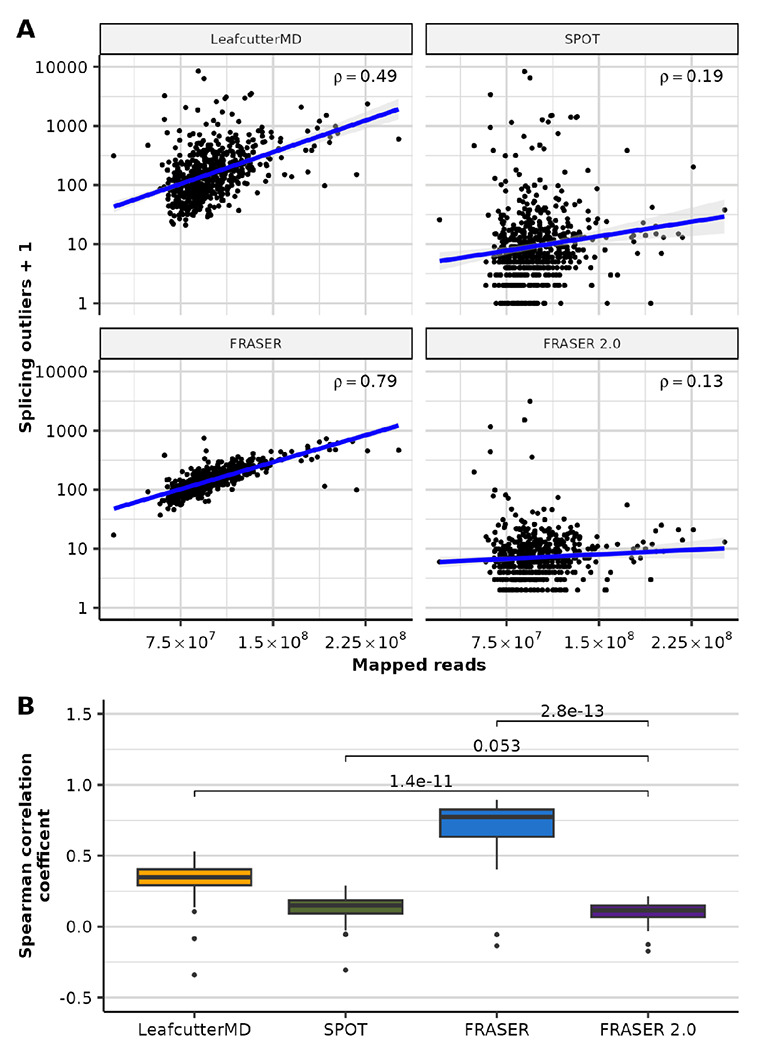
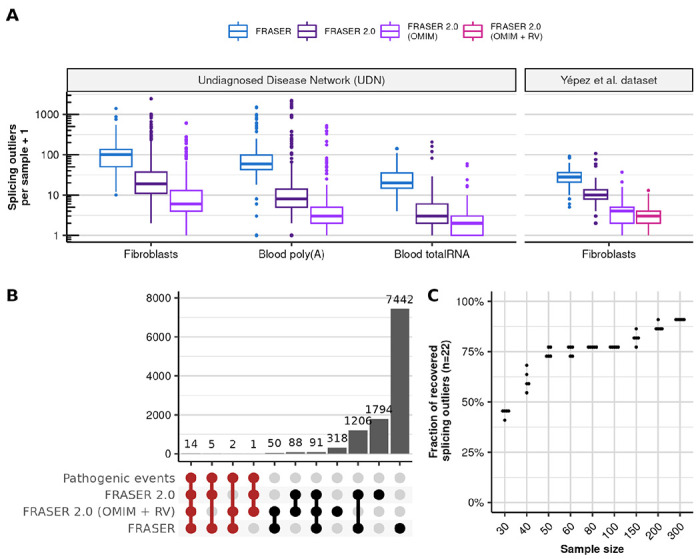
Detection of aberrantly spliced genes is an important step in RNA-seq-based rare disease diagnostics. We recently developed FRASER, a denoising autoencoder-based method for aberrant splicing detection that outperformed alternative approaches. However, as FRASER's three splice metrics are partially redundant and tend to be sensitive to sequencing depth, we introduce here a more robust intron excision metric, the Intron Jaccard Index, that combines alternative donor, alternative acceptor, and intron retention signal into a single value. Moreover, we optimized model parameters and filter cutoffs using candidate rare splice-disrupting variants as independent evidence. On 16,213 GTEx samples, our improved algorithm called typically 10 times fewer splicing outliers while increasing the proportion of candidate rare splice-disrupting variants by 10 fold and substantially decreasing the effect of sequencing depth on the number of reported outliers. Application on 303 rare disease samples confirmed the reduction fold-change of the number of outlier calls for a slight loss of sensitivity (only 2 out of 22 previously identified pathogenic splicing cases not recovered). Altogether, these methodological improvements contribute to more effective RNA-seq-based rare diagnostics by a drastic reduction of the amount of splicing outlier calls per sample at minimal loss of sensitivity.
Figures
References
Publication types
Grants and funding
- U01 HG007690/HG/NHGRI NIH HHS/United States
- U54 NS108251/NS/NINDS NIH HHS/United States
- U01 HG010219/HG/NHGRI NIH HHS/United States
- U01 HG007674/HG/NHGRI NIH HHS/United States
- U01 HG007672/HG/NHGRI NIH HHS/United States
- U01 HG010233/HG/NHGRI NIH HHS/United States
- U01 HG010230/HG/NHGRI NIH HHS/United States
- U01 HG007943/HG/NHGRI NIH HHS/United States
- U01 HG010217/HG/NHGRI NIH HHS/United States
- U01 HG007942/HG/NHGRI NIH HHS/United States
- U01 HG010215/HG/NHGRI NIH HHS/United States
- U01 HG007708/HG/NHGRI NIH HHS/United States
- U01 TR001395/TR/NCATS NIH HHS/United States
- U01 HG007709/HG/NHGRI NIH HHS/United States
- U54 NS093793/NS/NINDS NIH HHS/United States
- U01 HG007530/HG/NHGRI NIH HHS/United States
- U01 TR002471/TR/NCATS NIH HHS/United States
- U01 HG007703/HG/NHGRI NIH HHS/United States
LinkOut - more resources
Full Text Sources