

SOX17 Enhancer Variants Disrupt Transcription Factor Binding And Enhancer Inactivity Drives Pulmonary Hypertension
- PMID: 37066790
- PMCID: PMC7614572
- DOI: 10.1161/CIRCULATIONAHA.122.061940
SOX17 Enhancer Variants Disrupt Transcription Factor Binding And Enhancer Inactivity Drives Pulmonary Hypertension
Abstract
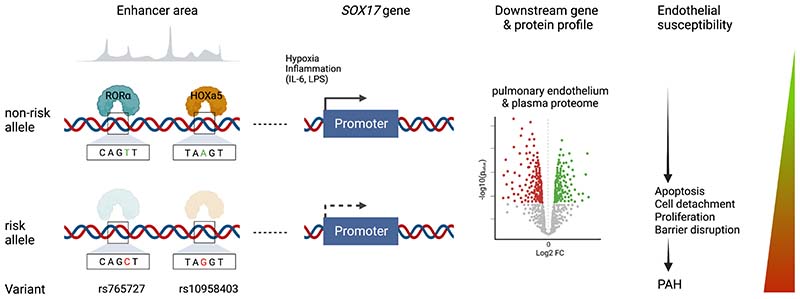
Background: Pulmonary arterial hypertension (PAH) is a rare disease characterized by remodeling of the pulmonary arteries, increased vascular resistance, and right-sided heart failure. Genome-wide association studies of idiopathic/heritable PAH established novel genetic risk variants, including conserved enhancers upstream of transcription factor (TF) SOX17 containing 2 independent signals. SOX17 is an important TF in embryonic development and in the homeostasis of pulmonary artery endothelial cells (hPAEC) in the adult. Rare pathogenic mutations in SOX17 cause heritable PAH. We hypothesized that PAH risk alleles in an enhancer region impair TF-binding upstream of SOX17, which in turn reduces SOX17 expression and contributes to disturbed endothelial cell function and PAH development.
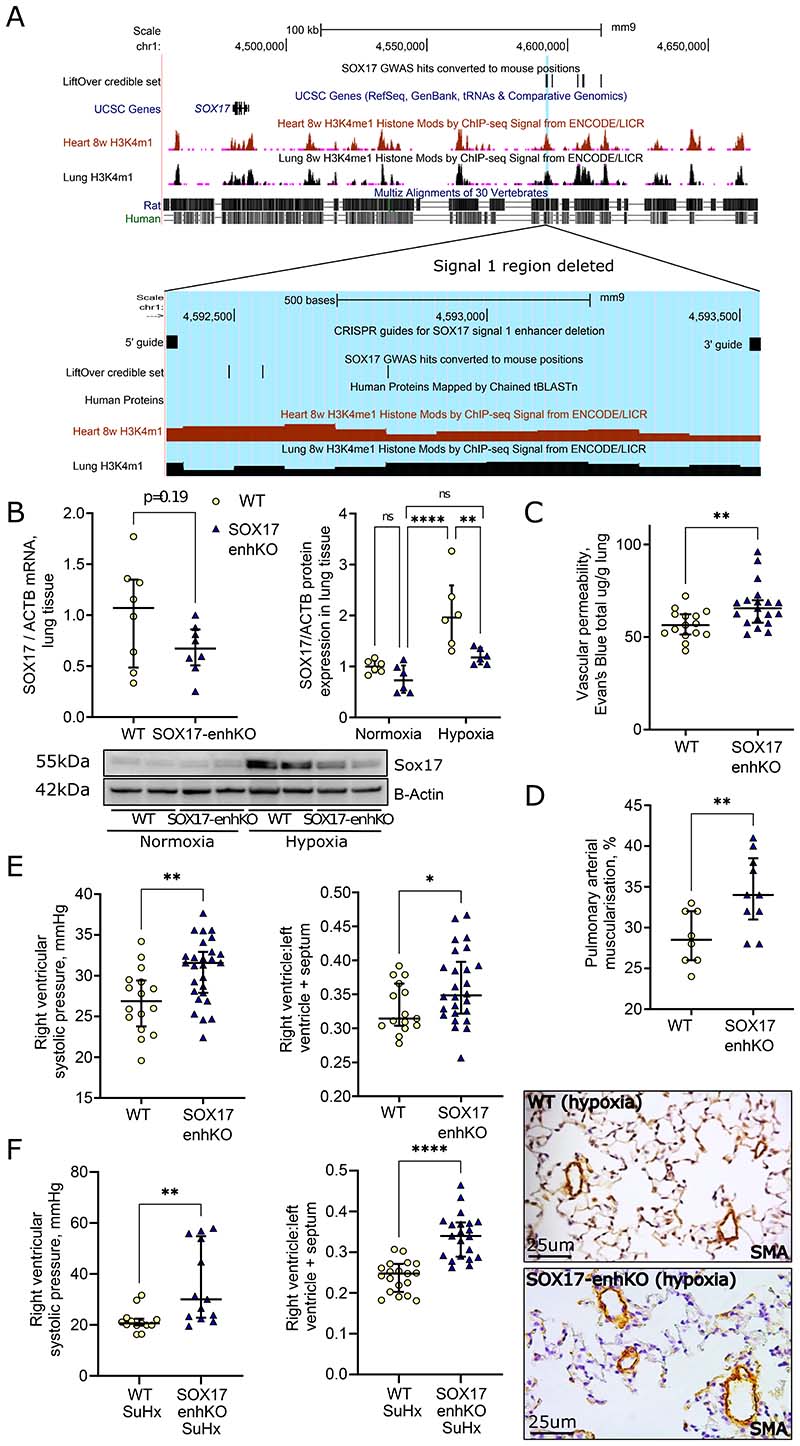
Methods: CRISPR manipulation and siRNA were used to modulate SOX17 expression. Electromobility shift assays were used to confirm in silico-predicted TF differential binding to the SOX17 variants. Functional assays in hPAECs were used to establish the biological consequences of SOX17 loss. In silico analysis with the connectivity map was used to predict compounds that rescue disturbed SOX17 signaling. Mice with deletion of the SOX17-signal 1 enhancer region (SOX17-4593/enhKO) were phenotyped in response to chronic hypoxia and SU5416/hypoxia.
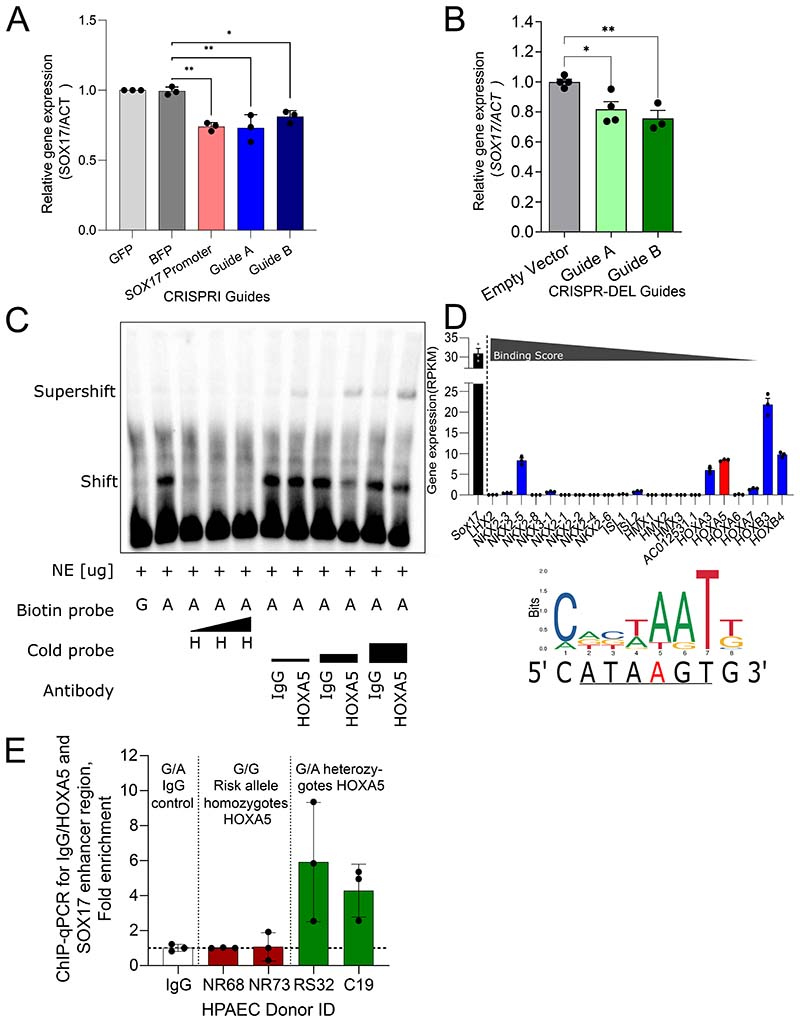
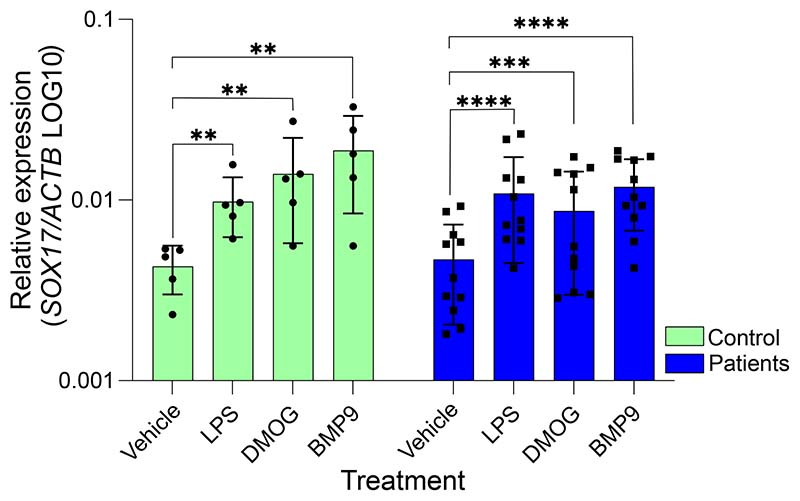
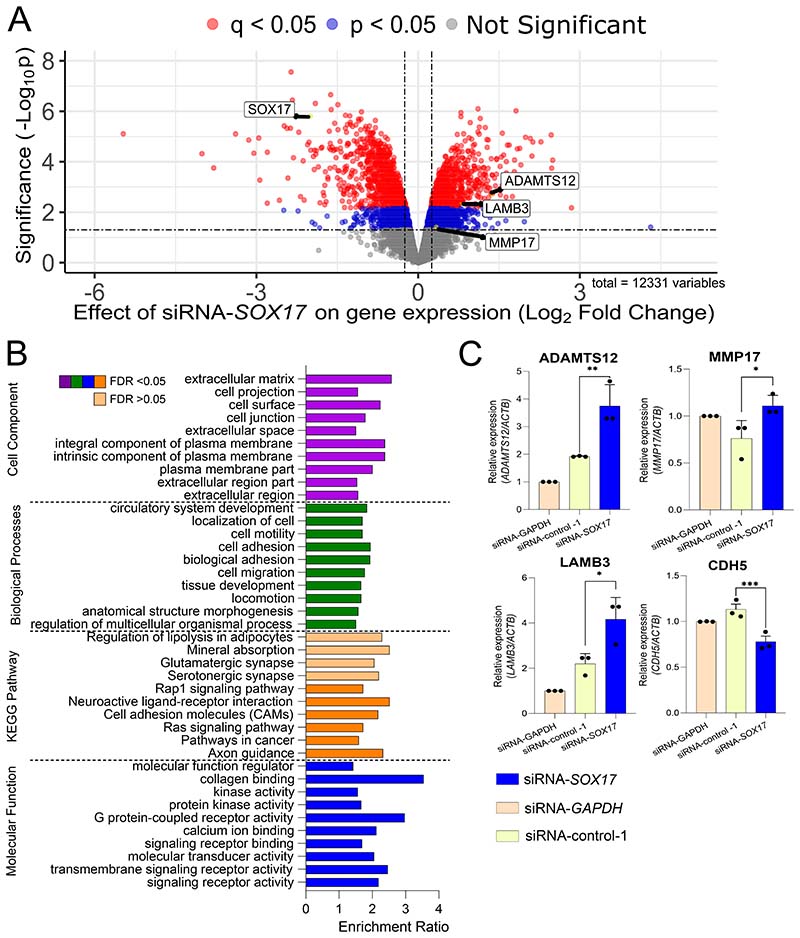
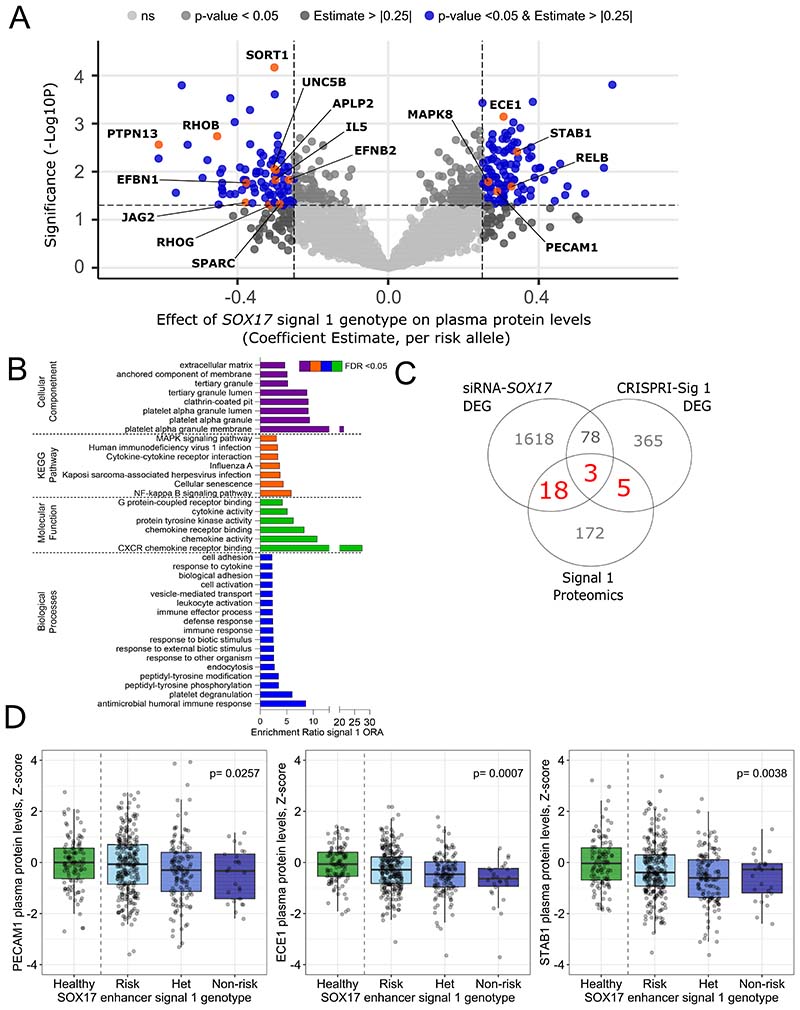
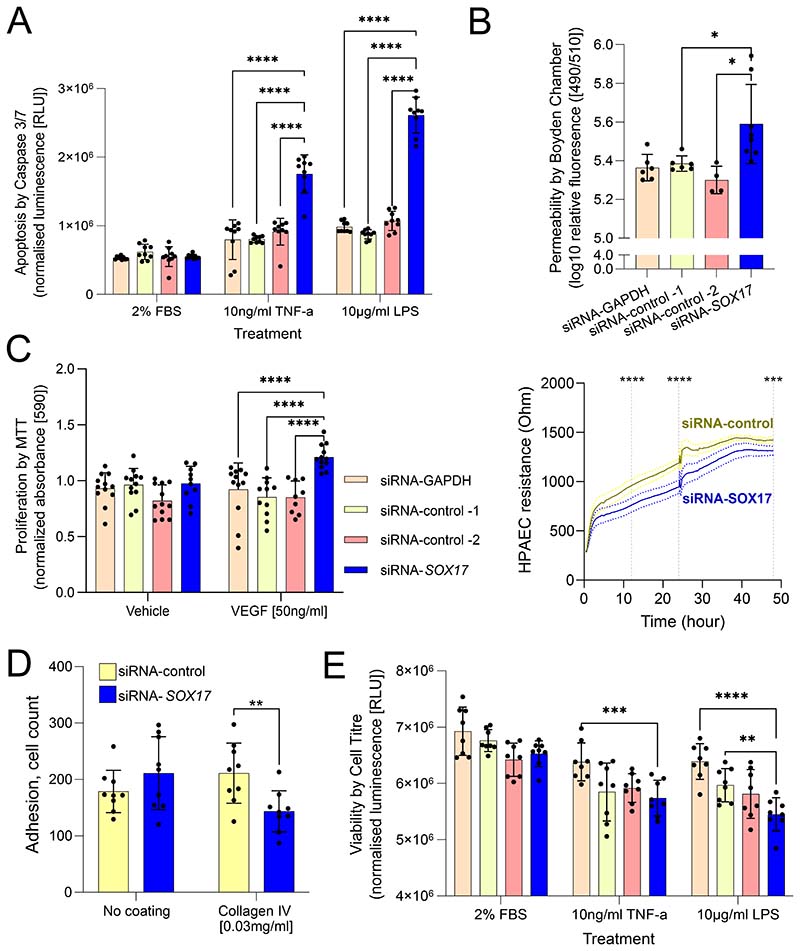
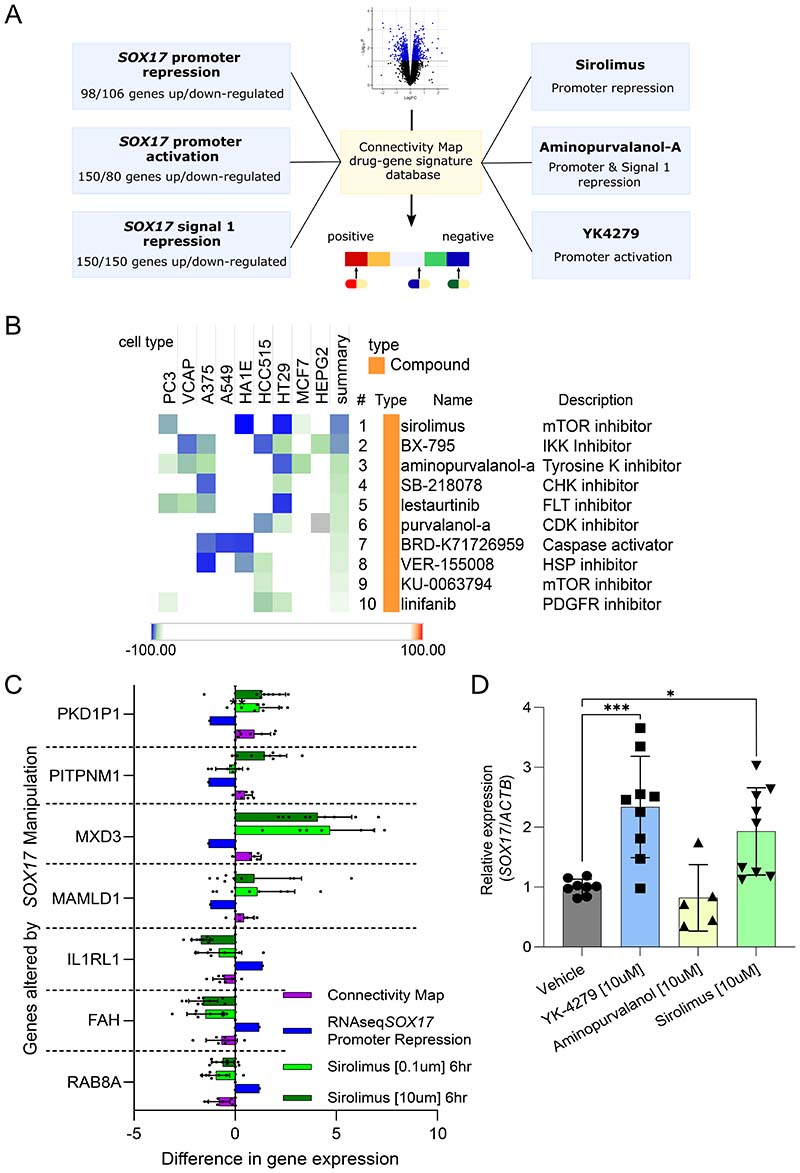
Results: CRISPR inhibition of SOX17-signal 2 and deletion of SOX17-signal 1 specifically decreased SOX17 expression. Electromobility shift assays demonstrated differential binding of hPAEC nuclear proteins to the risk and nonrisk alleles from both SOX17 signals. Candidate TFs HOXA5 and ROR-α were identified through in silico analysis and antibody electromobility shift assays. Analysis of the hPAEC transcriptomes revealed alteration of PAH-relevant pathways on SOX17 silencing, including extracellular matrix regulation. SOX17 silencing in hPAECs resulted in increased apoptosis, proliferation, and disturbance of barrier function. With the use of the connectivity map, compounds were identified that reversed the SOX17-dysfunction transcriptomic signatures in hPAECs. SOX17 enhancer knockout in mice reduced lung SOX17 expression, resulting in more severe pulmonary vascular leak and hypoxia or SU5416/hypoxia-induced pulmonary hypertension.
Conclusions: Common PAH risk variants upstream of the SOX17 promoter reduce endothelial SOX17 expression, at least in part, through differential binding of HOXA5 and ROR-α. Reduced SOX17 expression results in disturbed hPAEC function and PAH. Existing drug compounds can reverse the disturbed SOX17 pulmonary endothelial transcriptomic signature.
Keywords: SOX17 protein; human; hypertension; pulmonary.
Conflict of interest statement
Figures
Comment in
-
Letter by Li et al Regarding Article, "SOX17 Enhancer Variants Disrupt Transcription Factor Binding and Enhancer Inactivity Drives Pulmonary Hypertension".Circulation. 2023 Nov 28;148(22):1823-1824. doi: 10.1161/CIRCULATIONAHA.123.066230. Epub 2023 Nov 27. Circulation. 2023. PMID: 38011241 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
- FS/15/59/31839/BHF_/British Heart Foundation/United Kingdom
- SP/12/12/29836/BHF_/British Heart Foundation/United Kingdom
- P20 GM119943/GM/NIGMS NIH HHS/United States
- PG/19/17/34275/BHF_/British Heart Foundation/United Kingdom
- MR/K020919/1/MRC_/Medical Research Council/United Kingdom
- 101033/WT_/Wellcome Trust/United Kingdom
- MR/L02036X/1/MRC_/Medical Research Council/United Kingdom
- FS/SBSRF/22/31025/BHF_/British Heart Foundation/United Kingdom
- SP/18/10/33975/BHF_/British Heart Foundation/United Kingdom
- 789055/ERC_/European Research Council/International
- R01 HL158841/HL/NHLBI NIH HHS/United States
- RE/18/4/34215/BHF_/British Heart Foundation/United Kingdom
- PG/18/2/33446/BHF_/British Heart Foundation/United Kingdom
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
