

Multivariate genome-wide association analysis by iterative hard thresholding
- PMID: 37067496
- PMCID: PMC10133532
- DOI: 10.1093/bioinformatics/btad193
Multivariate genome-wide association analysis by iterative hard thresholding
Abstract
Motivation: In a genome-wide association study, analyzing multiple correlated traits simultaneously is potentially superior to analyzing the traits one by one. Standard methods for multivariate genome-wide association study operate marker-by-marker and are computationally intensive.
Results: We present a sparsity constrained regression algorithm for multivariate genome-wide association study based on iterative hard thresholding and implement it in a convenient Julia package MendelIHT.jl. In simulation studies with up to 100 quantitative traits, iterative hard thresholding exhibits similar true positive rates, smaller false positive rates, and faster execution times than GEMMA's linear mixed models and mv-PLINK's canonical correlation analysis. On UK Biobank data with 470 228 variants, MendelIHT completed a three-trait joint analysis (n=185 656) in 20 h and an 18-trait joint analysis (n=104 264) in 53 h with an 80 GB memory footprint. In short, MendelIHT enables geneticists to fit a single regression model that simultaneously considers the effect of all SNPs and dozens of traits.
Availability and implementation: Software, documentation, and scripts to reproduce our results are available from https://github.com/OpenMendel/MendelIHT.jl.
© The Author(s) 2023. Published by Oxford University Press.
Conflict of interest statement
None declared.
Figures
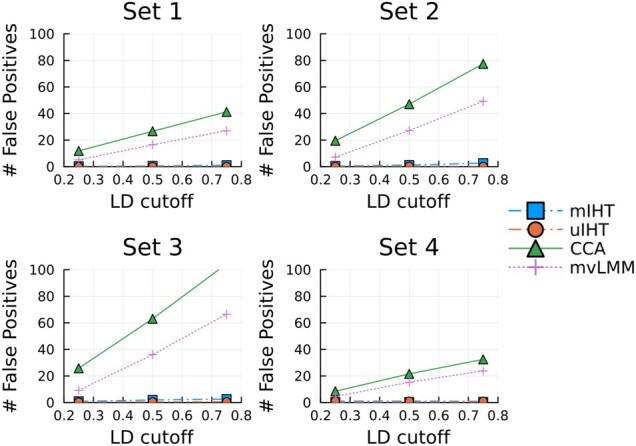
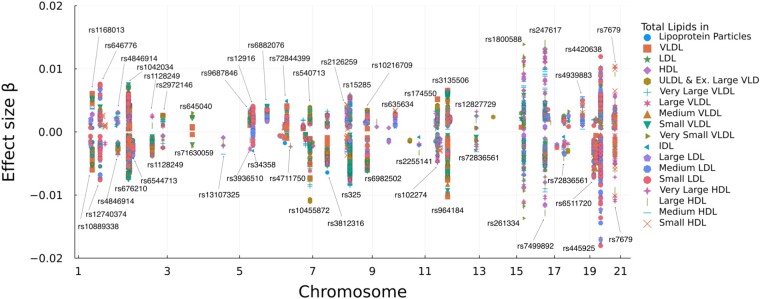
References
-
- Abraham G, Qiu Y, Inouye M. et al. FlashPCA2: principal component analysis of Biobank-scale genotype datasets. Bioinformatics 2017;33:2776–8. - PubMed
-
- Alexander DH, Lange K.. Stability selection for genome-wide association. Genet Epidemiol 2011;35:722–8. - PubMed
-
- Barber RF, Candès EJ.. Controlling the false discovery rate via knockoffs. Ann Statist 2015;43:2055–85.
-
- Bezanson J, Edelman A, Karpinski S. et al. Julia: a fresh approach to numerical computing. SIAM Rev 2017;59:65–98.
