

Li-Fraumeni Syndrome-Associated Dimer-Forming Mutant p53 Promotes Transactivation-Independent Mitochondrial Cell Death
- PMID: 37067901
- PMCID: PMC10287063
- DOI: 10.1158/2159-8290.CD-22-0882
Li-Fraumeni Syndrome-Associated Dimer-Forming Mutant p53 Promotes Transactivation-Independent Mitochondrial Cell Death
Abstract
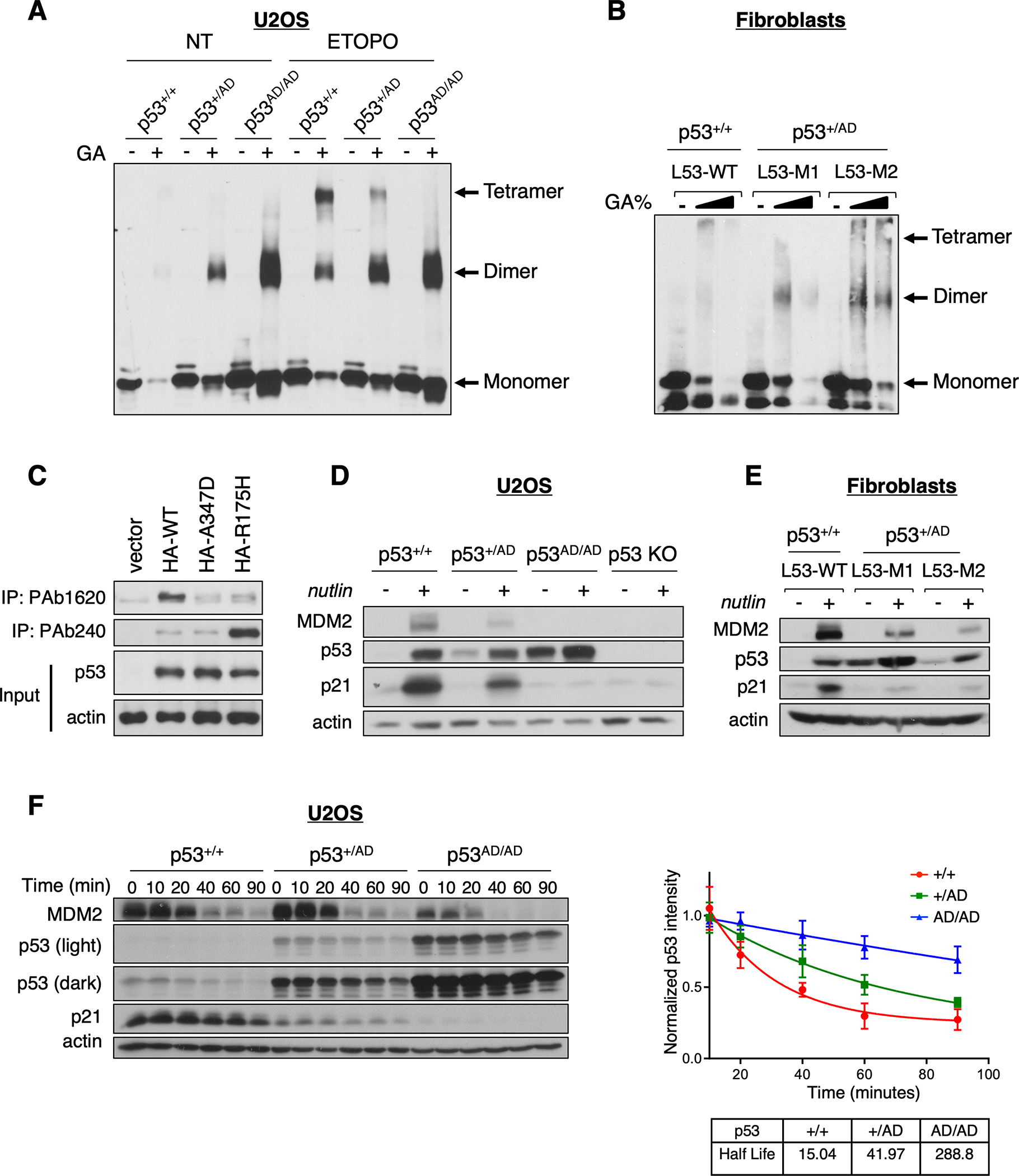
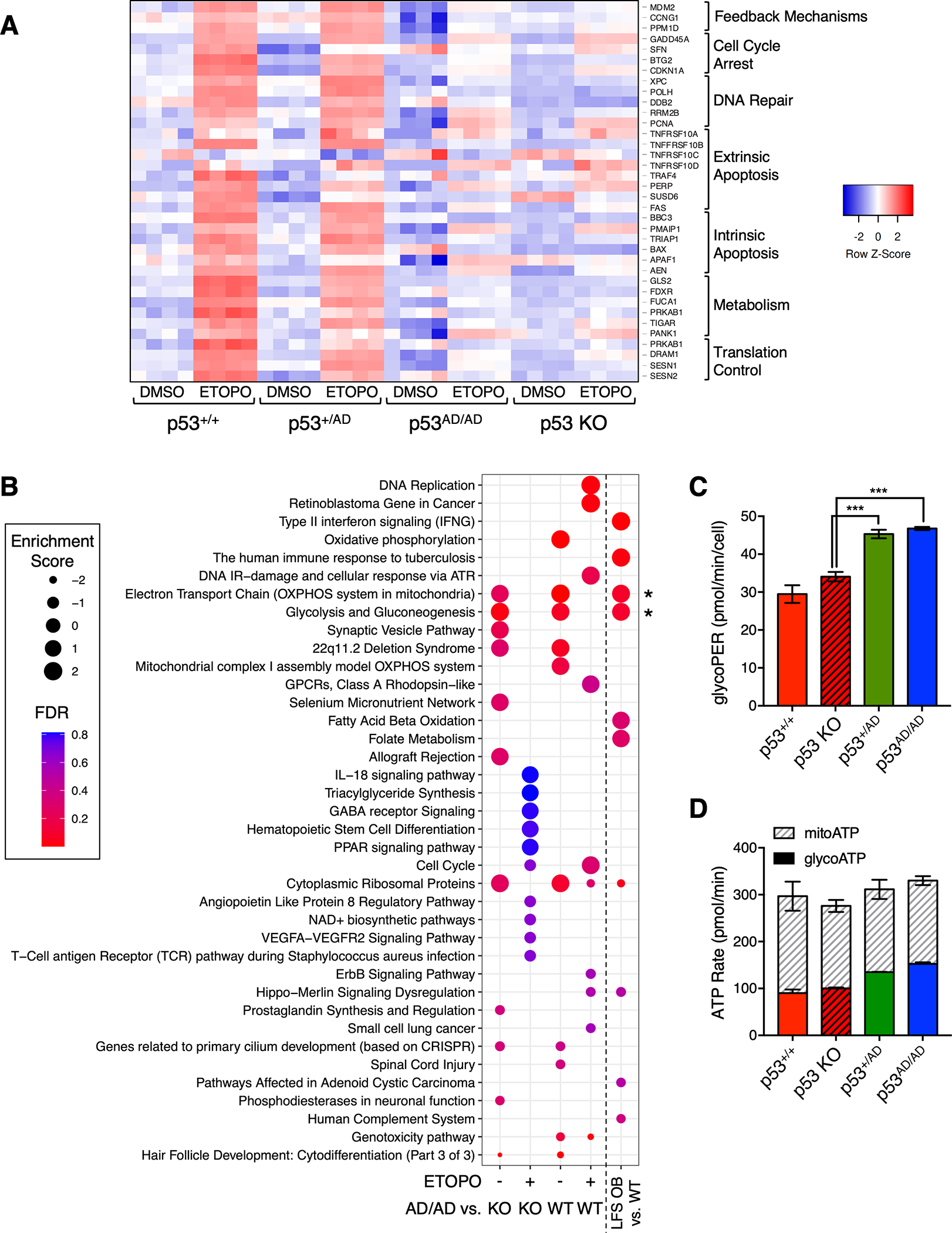
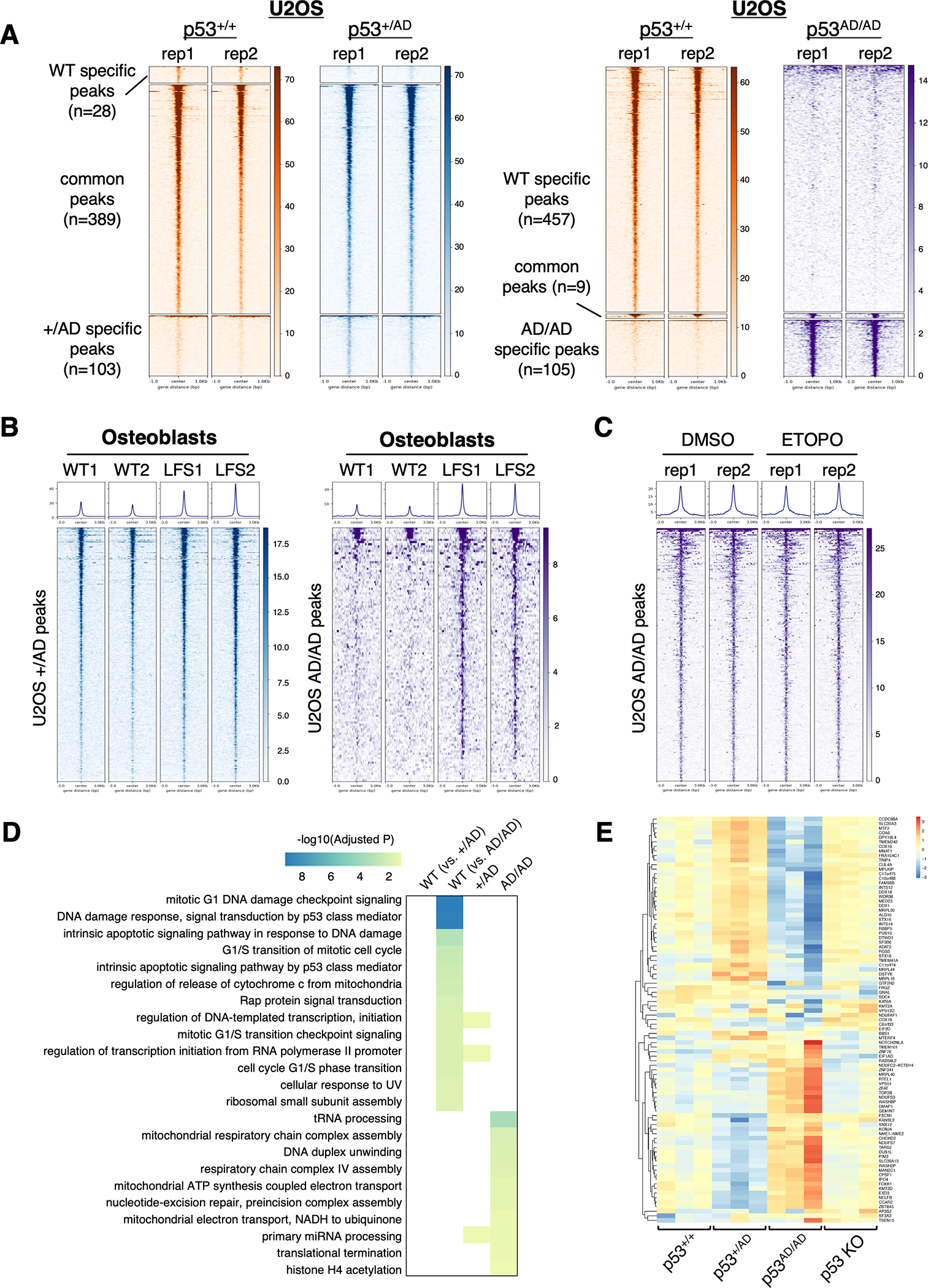
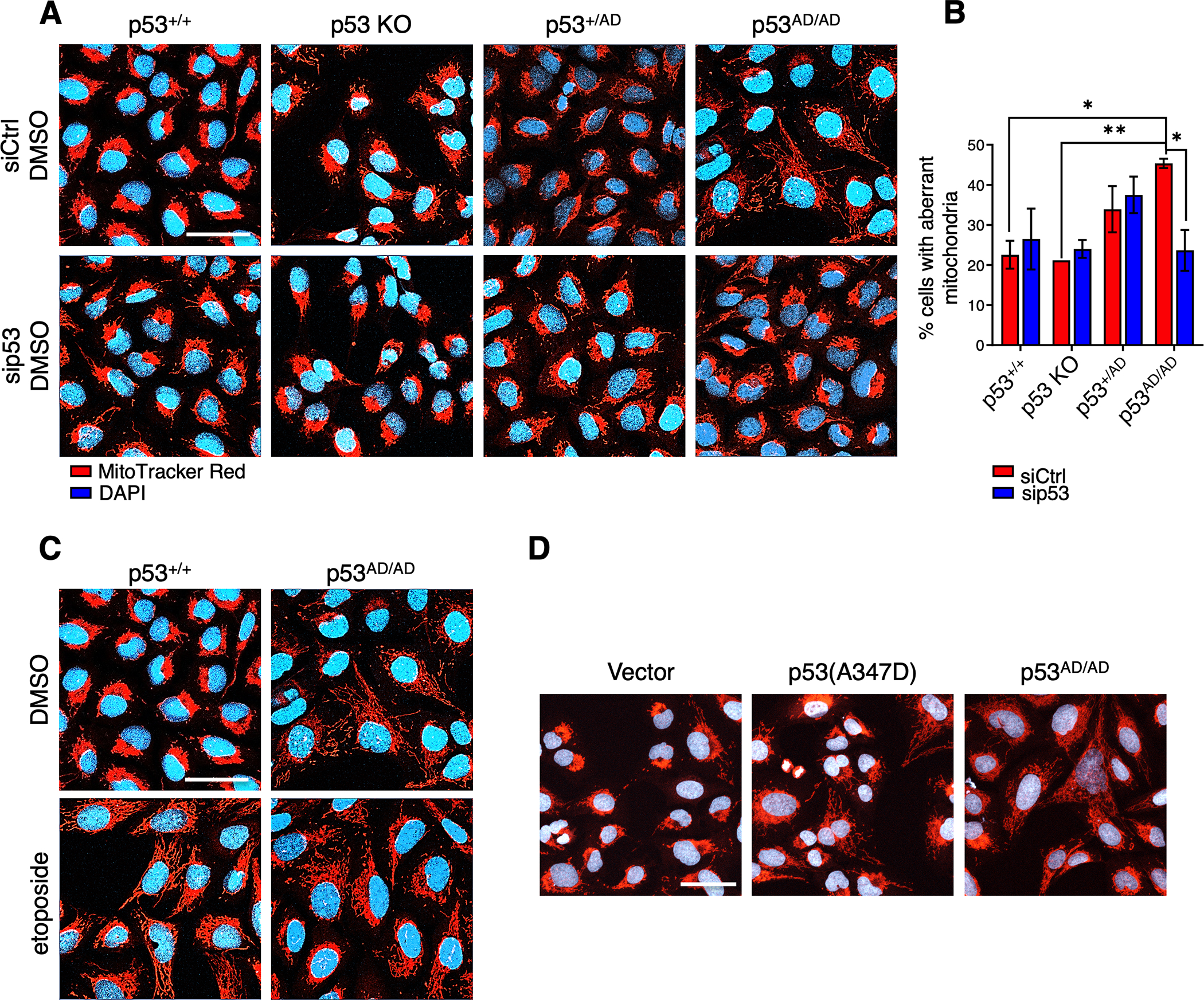
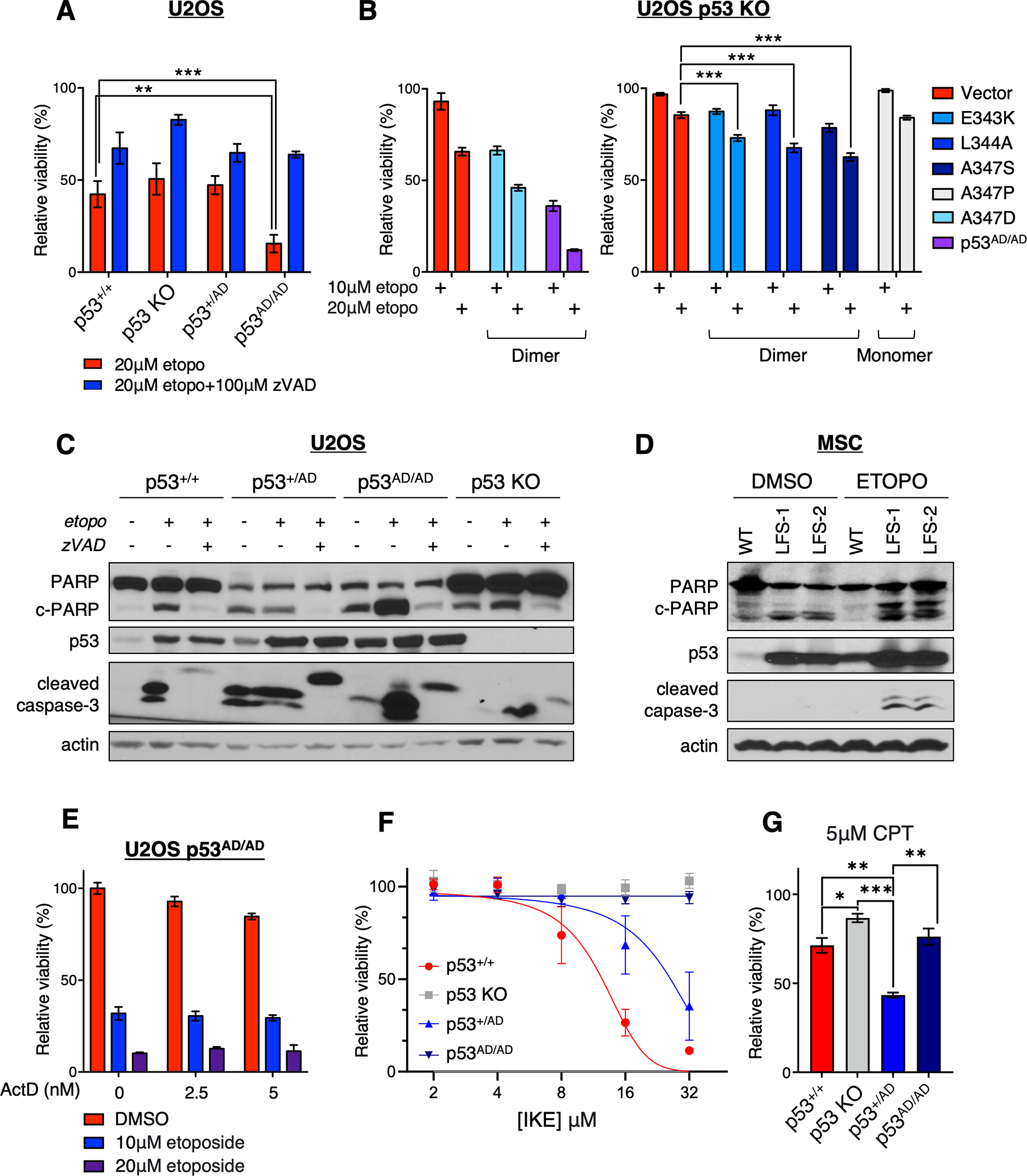
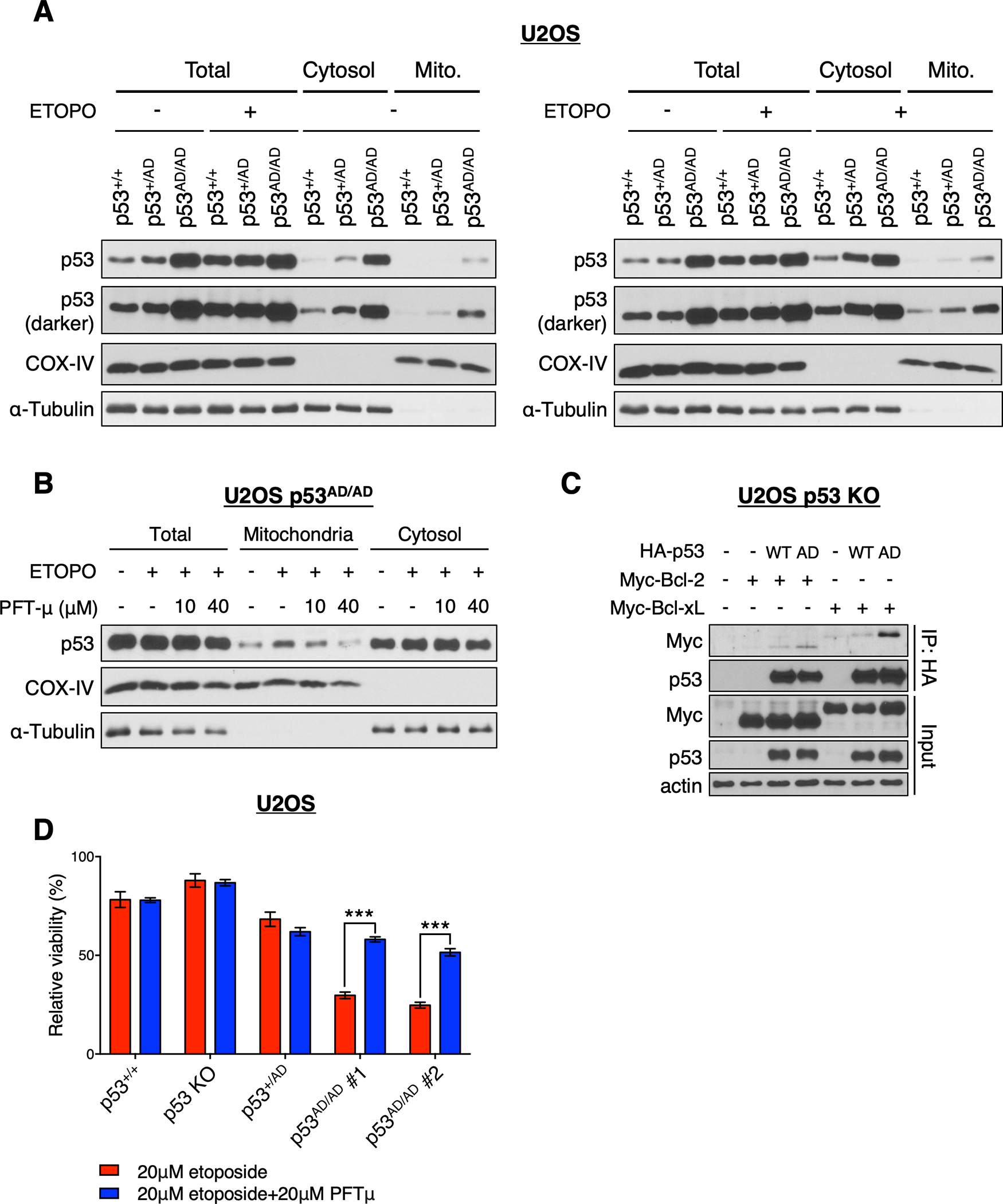
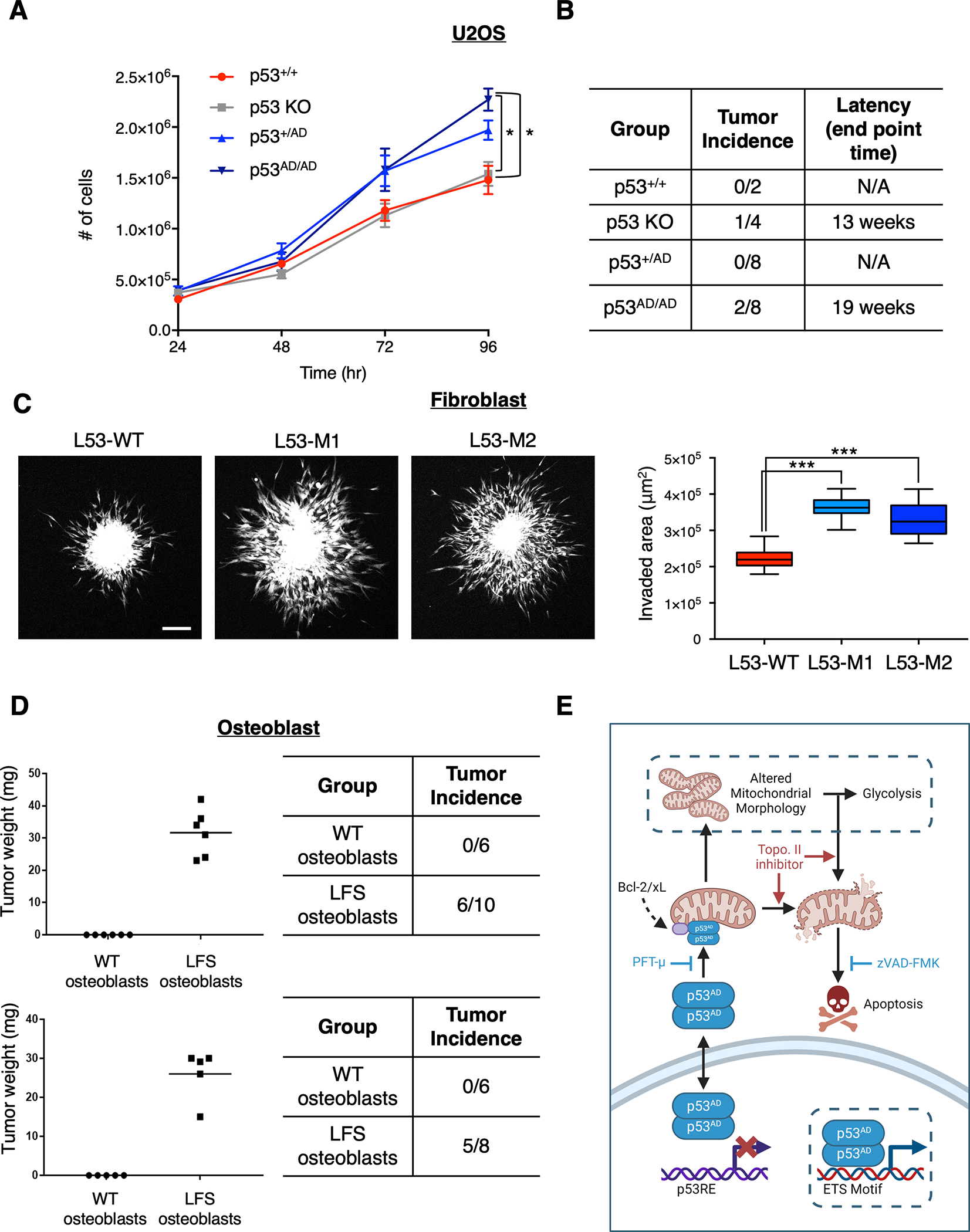
Cancer-relevant mutations in the oligomerization domain (OD) of the p53 tumor suppressor protein, unlike those in the DNA binding domain, have not been well elucidated. Here, we characterized the germline OD mutant p53(A347D), which occurs in cancer-prone Li-Fraumeni syndrome (LFS) patients. Unlike wild-type p53, mutant p53(A347D) cannot form tetramers and exists as a hyperstable dimeric protein. Further, p53(A347D) cannot bind or transactivate the majority of canonical p53 target genes. Isogenic cell lines harboring either p53(A347D) or no p53 yield comparable tumorigenic properties, yet p53(A347D) displays remarkable neomorphic activities. Cells bearing p53(A347D) possess a distinct transcriptional profile and undergo metabolic reprogramming. Further, p53(A347D) induces striking mitochondrial network aberration and associates with mitochondria to drive apoptotic cell death upon topoisomerase II inhibition in the absence of transcription. Thus, dimer-forming p53 demonstrates both loss-of-function (LOF) and gain-of-function (GOF) properties compared with the wild-type form of the protein.
Significance: A mutant p53 (A347D), which can only form dimers, is associated with increased cancer susceptibility in LFS individuals. We found that this mutant wields a double-edged sword, driving tumorigenesis through LOF while gaining enhanced apoptogenic activity as a new GOF, thereby yielding a potential vulnerability to select therapeutic approaches. See related commentary by Stieg et al., p. 1046. See related article by Gencel-Augusto et al., p. 1230. This article is highlighted in the In This Issue feature, p. 1027.
©2023 American Association for Cancer Research.
Conflict of interest statement
Conflict of Interest Statement:
The authors declare no conflicts with the research conducted in this study.
Figures
Comment in
-
Dimeric p53 Mutant Elicits Unique Tumor-Suppressive Activities through an Altered Metabolic Program.Cancer Discov. 2023 May 4;13(5):1230-1249. doi: 10.1158/2159-8290.CD-22-0872. Cancer Discov. 2023. PMID: 37067911 Free PMC article.
-
p53 Oligomerization Domain Mutants: A New Class of Mutants That Retain "License to Kill".Cancer Discov. 2023 May 4;13(5):1046-1048. doi: 10.1158/2159-8290.CD-23-0200. Cancer Discov. 2023. PMID: 37139723
Comment on
-
Dimeric p53 Mutant Elicits Unique Tumor-Suppressive Activities through an Altered Metabolic Program.Cancer Discov. 2023 May 4;13(5):1230-1249. doi: 10.1158/2159-8290.CD-22-0872. Cancer Discov. 2023. PMID: 37067911 Free PMC article.
References
-
- Hollstein M, Sidransky D, Vogelstein B, Harris CC. p53 mutations in human cancers. Science 1991;253(5015):49–53. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
