

NF-κB is a critical mediator of post-mitotic senescence in oligodendrocytes and subsequent white matter loss
- PMID: 37069623
- PMCID: PMC10108549
- DOI: 10.1186/s13024-023-00616-5
NF-κB is a critical mediator of post-mitotic senescence in oligodendrocytes and subsequent white matter loss
Abstract
Background: Inflammaging represents an accepted concept where the immune system shifts to a low-grade chronic pro-inflammatory state without overt infection upon aging. In the CNS, inflammaging is mainly driven by glia cells and associated with neurodegenerative processes. White matter degeneration (WMD), a well-known process in the aging brain, manifests in myelin loss finally resulting in motor, sensory and cognitive impairments. Oligodendrocytes (OL) are responsible for homeostasis and maintenance of the myelin sheaths, which is a complex and highly energy demanding process sensitizing these cells to metabolic, oxidative and other forms of stress. Yet, the immediate impact of chronic inflammatory stress like inflammaging on OL homeostasis, myelin maintenance and WMD remains open.
Methods: To functionally analyze the role of IKK/NF-κB signaling in the regulation of myelin homeostasis and maintenance in the adult CNS, we established a conditional mouse model allowing NF-κB activation in mature myelinating oligodendrocytes. IKK2-CAPLP-CreERT2 mice were characterized by biochemical, immunohistochemical, ultrastructural and behavioral analyses. Transcriptome data from isolated, primary OLs and microglia cells were explored by in silico pathway analysis and validated by complementary molecular approaches.
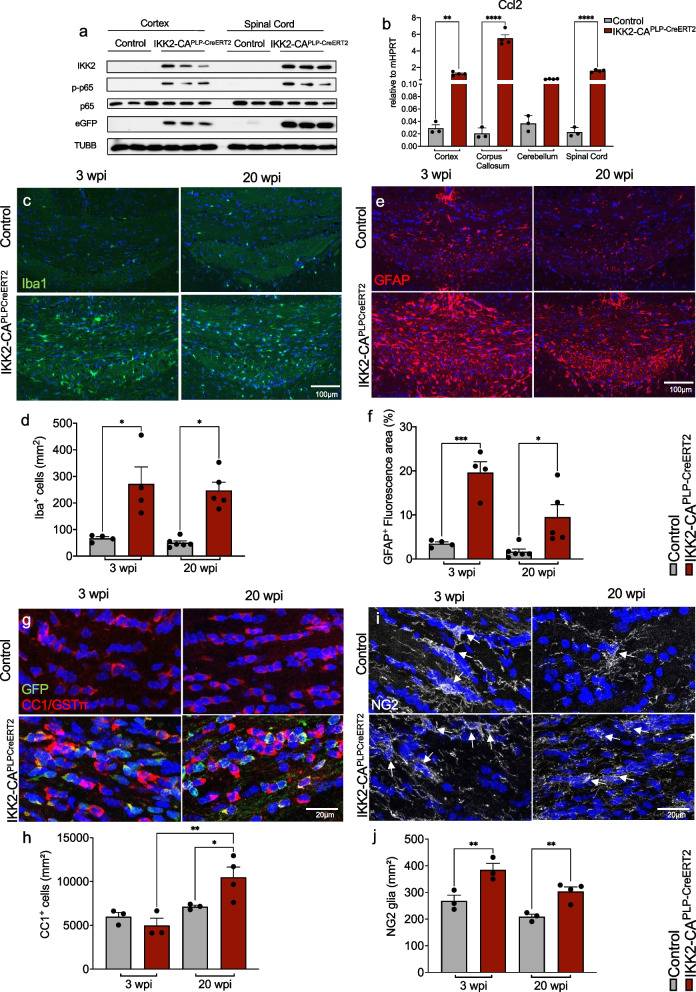
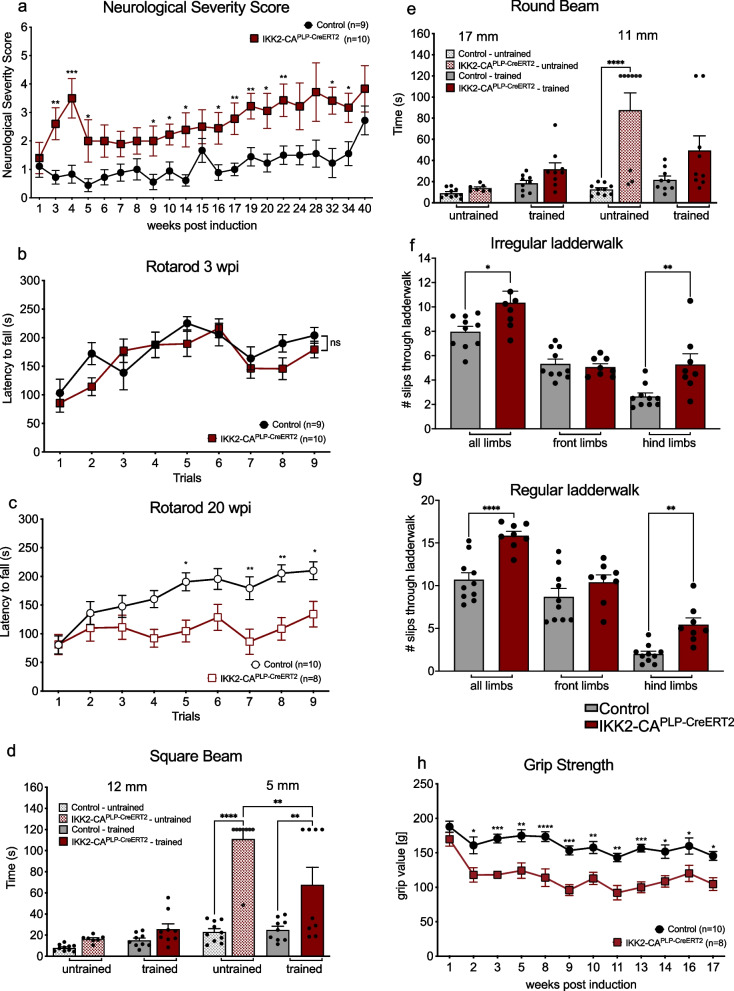
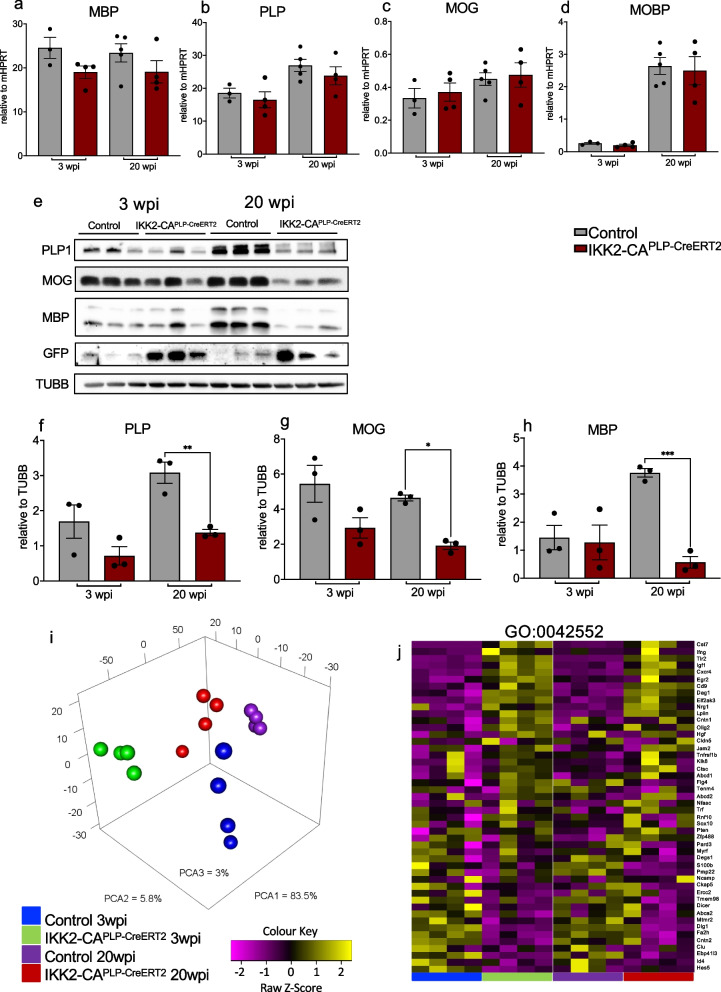
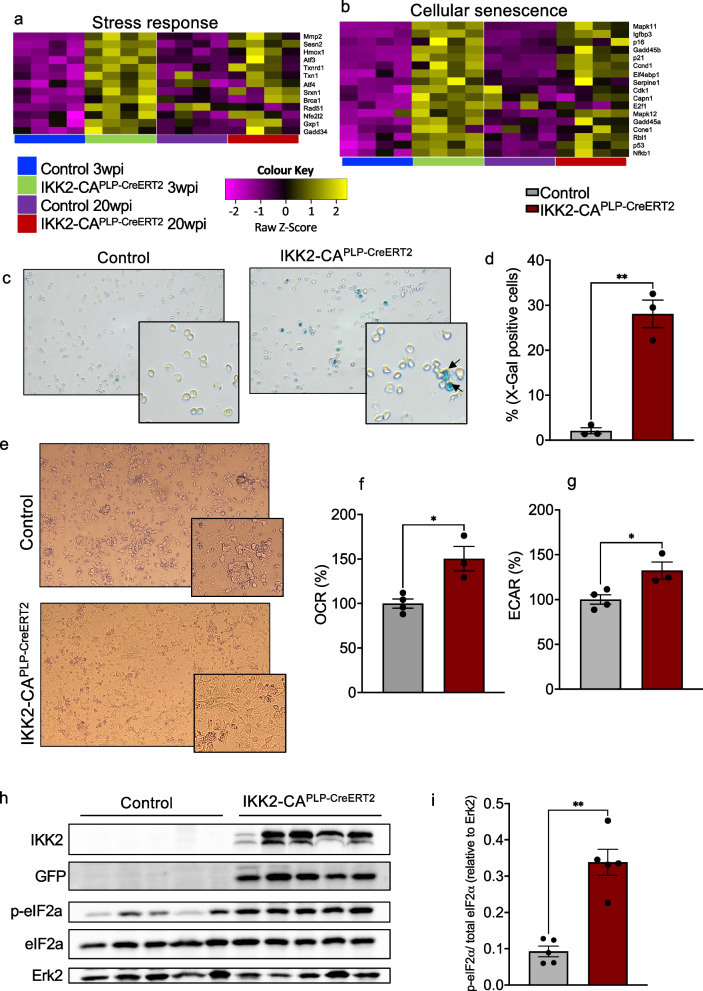
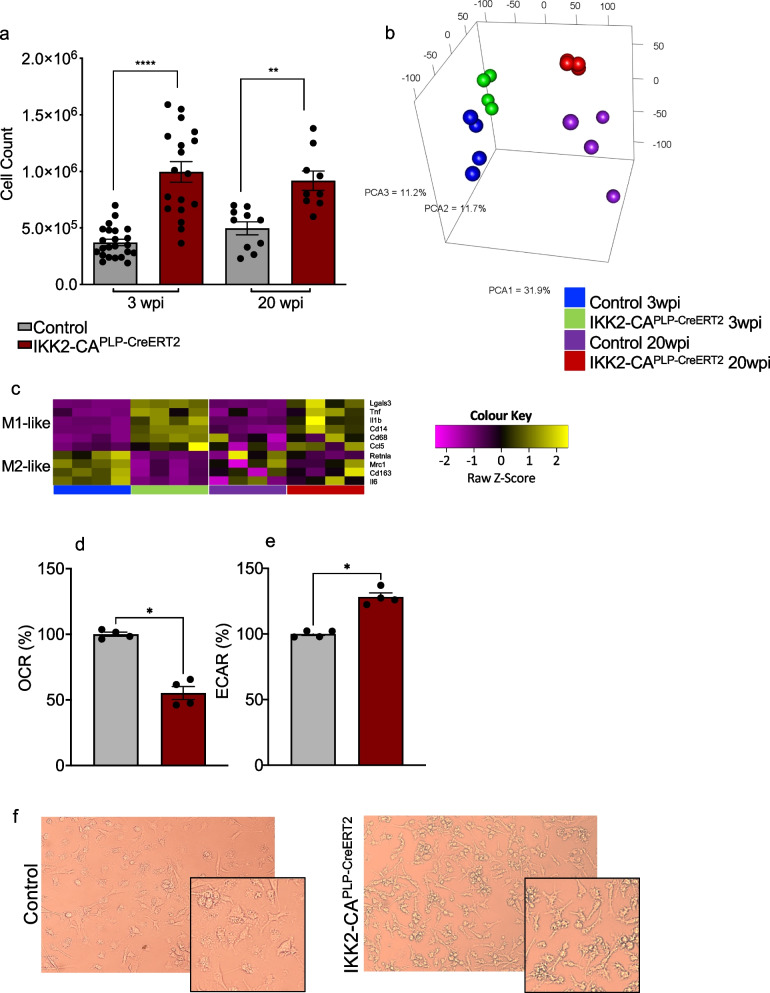
Results: Chronic NF-κB activation in mature OLs leads to aggravated neuroinflammatory conditions phenocopying brain inflammaging. As a consequence, IKK2-CAPLP-CreERT2 mice showed specific neurological deficits and impaired motoric learning. Upon aging, persistent NF-κB signaling promotes WMD in these mice as ultrastructural analysis revealed myelination deficits in the corpus callosum accompanied by impaired myelin protein expression. RNA-Seq analysis of primary oligodendrocytes and microglia cells uncovers gene expression signatures associated with activated stress responses and increased post mitotic cellular senescence (PoMiCS) which was confirmed by elevated senescence-associated β-galactosidase activity and SASP gene expression profile. We identified an elevated integrated stress response (ISR) characterized by phosphorylation of eIF2α as a relevant molecular mechanism which is able to affect translation of myelin proteins.
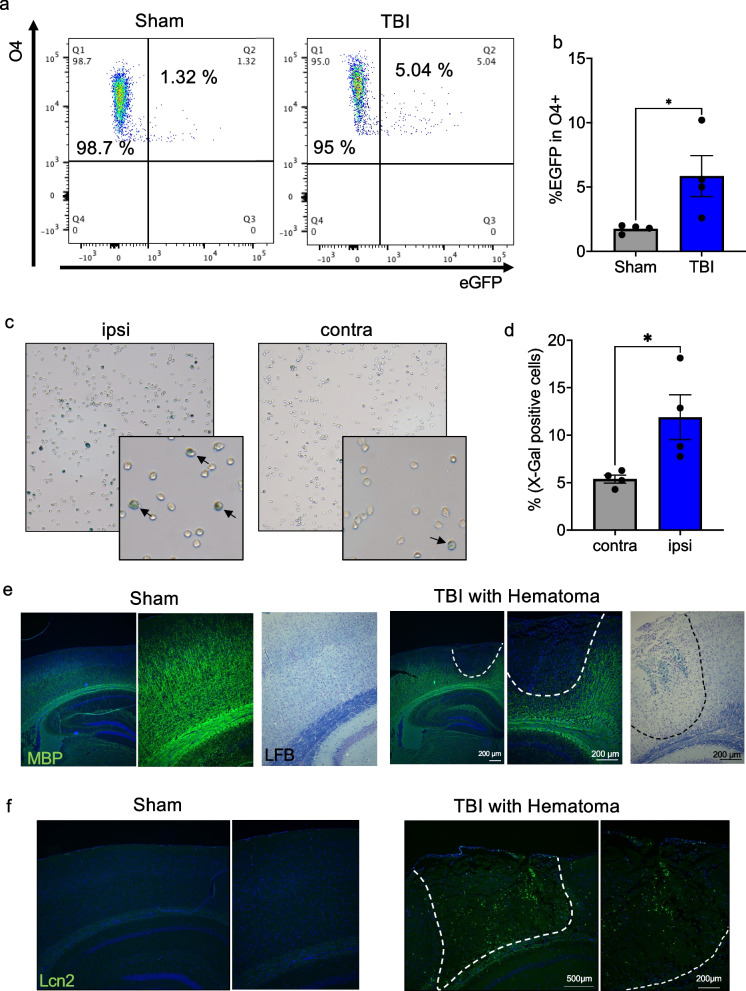
Conclusions: Our findings demonstrate an essential role of IKK/NF-κB signaling in mature, post-mitotic OLs in regulating stress-induced senescence in these cells. Moreover, our study identifies PoMICS as an important driving force of age-dependent WMD as well as of traumatic brain injury induced myelin defects.
Keywords: Inflammaging; Integrated stress response; Mature oligodendrocytes; NF-κB; PoMICS; White matter degeneration.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
Figures

References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
