

Consensus clinical management guidelines for acid sphingomyelinase deficiency (Niemann-Pick disease types A, B and A/B)
- PMID: 37069638
- PMCID: PMC10108815
- DOI: 10.1186/s13023-023-02686-6
Consensus clinical management guidelines for acid sphingomyelinase deficiency (Niemann-Pick disease types A, B and A/B)
Abstract
Background: Acid Sphingomyelinase Deficiency (ASMD) is a rare autosomal recessive disorder caused by mutations in the SMPD1 gene. This rarity contributes to misdiagnosis, delayed diagnosis and barriers to good care. There are no published national or international consensus guidelines for the diagnosis and management of patients with ASMD. For these reasons, we have developed clinical guidelines that defines standard of care for ASMD patients.
Methods: The information contained in these guidelines was obtained through a systematic literature review and the experiences of the authors in their care of patients with ASMD. We adopted the Appraisal of Guidelines for Research and Evaluation (AGREE II) system as method of choice for the guideline development process.
Results: The clinical spectrum of ASMD, although a continuum, varies substantially with subtypes ranging from a fatal infantile neurovisceral disorder to an adult-onset chronic visceral disease. We produced 39 conclusive statements and scored them according to level of evidence, strengths of recommendations and expert opinions. In addition, these guidelines have identified knowledge gaps that must be filled by future research.
Conclusion: These guidelines can inform care providers, care funders, patients and their carers about best clinical practice and leads to a step change in the quality of care for patients with ASMD with or without enzyme replacement therapy (ERT).
Keywords: ASMD; Acid sphingomyelinase deficiency; Diagnosis; Guidelines; Management; Niemann–Pick disease; Niemann–Pick disease-a,b,a/b.
© 2023. The Author(s).
Conflict of interest statement
Sanofi has provided financial support to AD, AL, ES, OL, RG, SCB and TH, research grant to ES and TH, and honorarium to AD, ES, OL, MTV and RG. Honorarium has been provided by Orphazyme to AD and MTV.
Figures
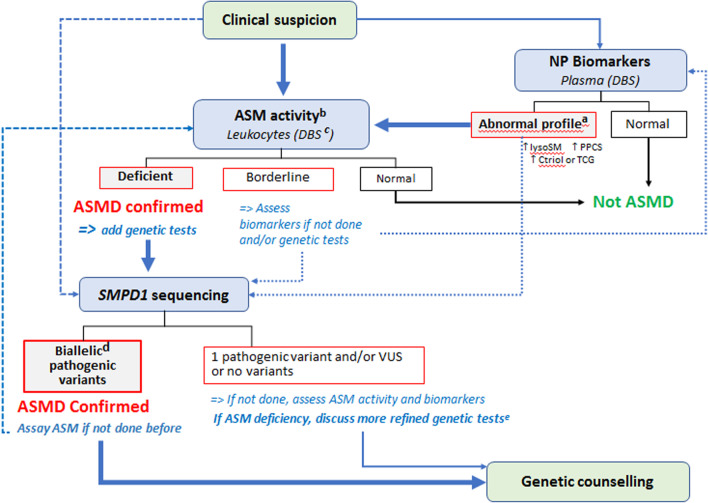
References
-
- Brouwers MC, Kho ME, Browman GP, Burgers JS, Cluzeau F, Feder G, Fervers B, Graham ID, Grimshaw J, Hanna SE, Littlejohns P, Makarski J, Zitzelsberger L; AGREE Next Steps Consortium. AGREE II: advancing guideline development, reporting and evaluation in health care. CMAJ. 2010;182(18):E839–42 - PMC - PubMed
-
- Landrum MJ, Lee JM, Benson M, Brown GR, Chao C, Chitipiralla S, Gu B, Hart J, Hoffman D, Jang W, Karapetyan K, Katz K, Liu C, Maddipatla Z, Malheiro A, McDaniel K, Ovetsky M, Riley G, Zhou G, Holmes JB, Kattman BL, Maglott DR. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018;46(D1):D1062–D1067. doi: 10.1093/nar/gkx1153. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
