

Transgressive and parental dominant gene expression and cytosine methylation during seed development in Brassica napus hybrids
- PMID: 37071201
- PMCID: PMC10113308
- DOI: 10.1007/s00122-023-04345-7
Transgressive and parental dominant gene expression and cytosine methylation during seed development in Brassica napus hybrids
Abstract
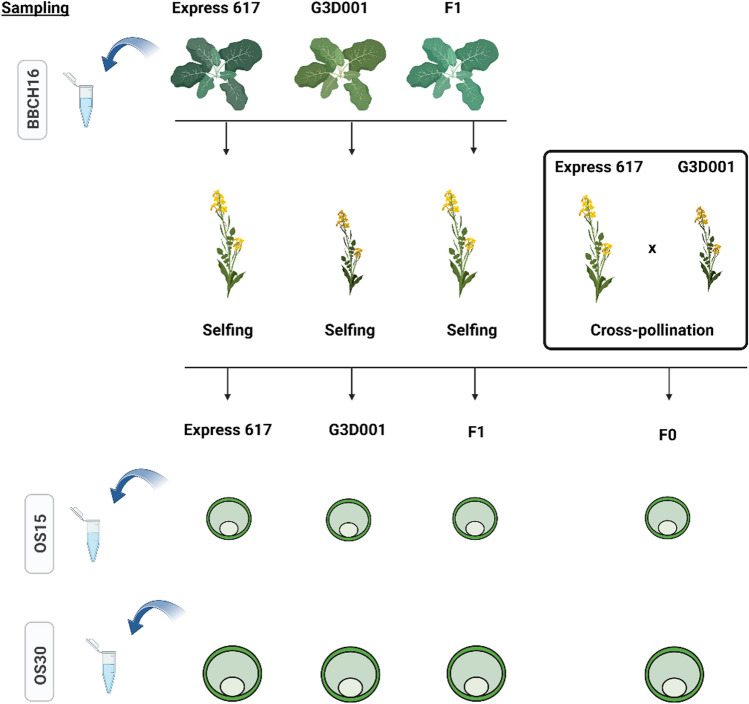
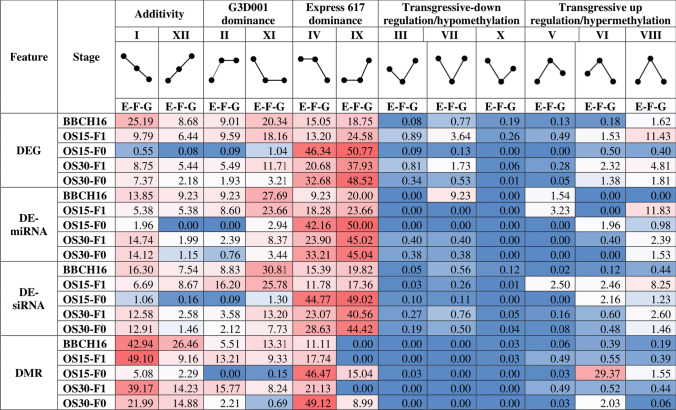
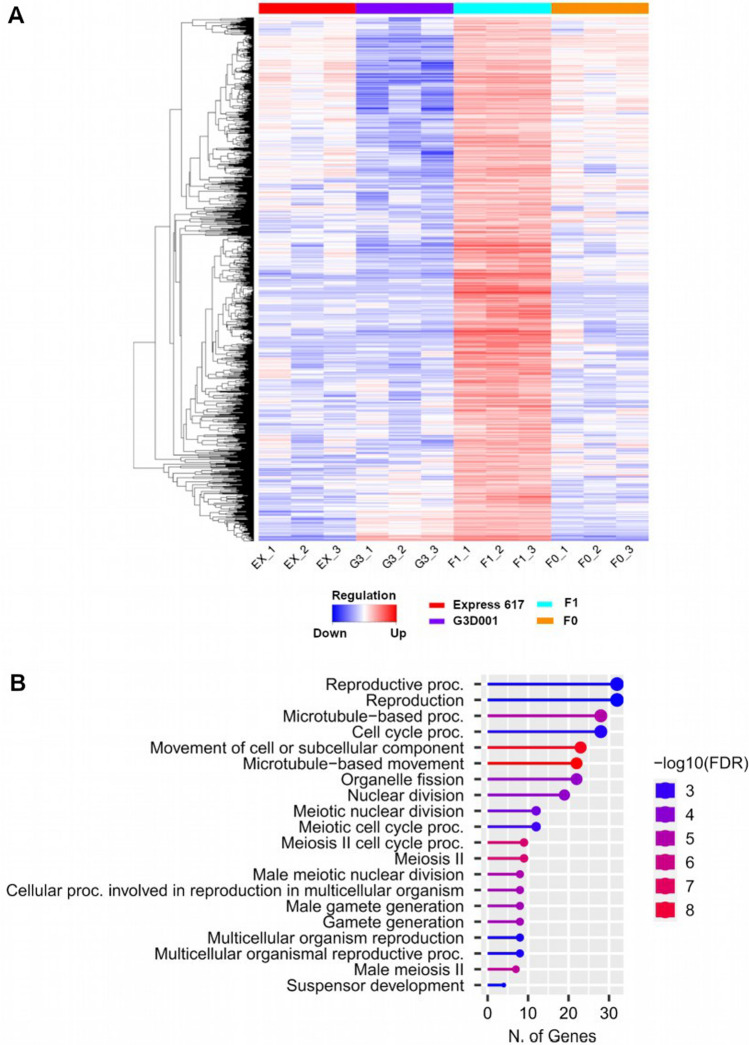
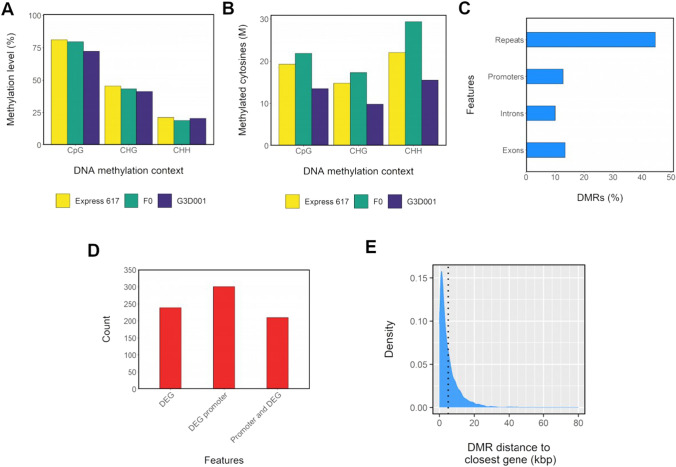
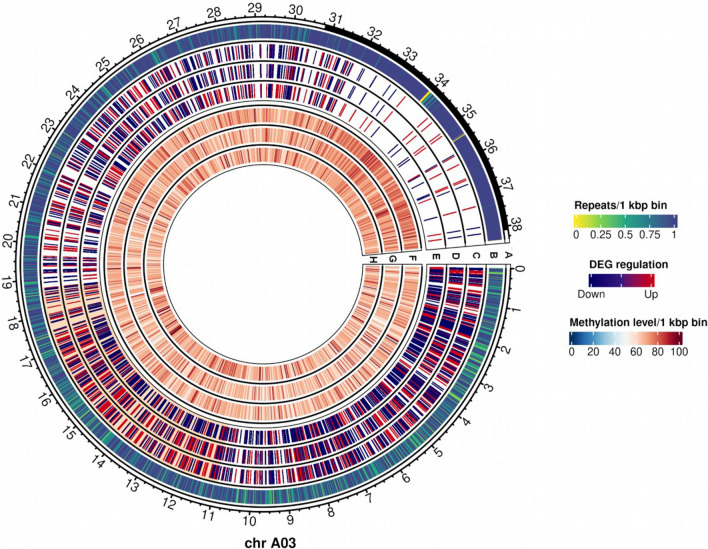
Transcriptomic and epigenomic profiling of gene expression and small RNAs during seed and seedling development reveals expression and methylation dominance levels with implications on early stage heterosis in oilseed rape. The enhanced performance of hybrids through heterosis remains a key aspect in plant breeding; however, the underlying mechanisms are still not fully elucidated. To investigate the potential role of transcriptomic and epigenomic patterns in early expression of hybrid vigor, we investigated gene expression, small RNA abundance and genome-wide methylation in hybrids from two distant Brassica napus ecotypes during seed and seedling developmental stages using next-generation sequencing. A total of 31117, 344, 36229 and 7399 differentially expressed genes, microRNAs, small interfering RNAs and differentially methylated regions were identified, respectively. Approximately 70% of the differentially expressed or methylated features displayed parental dominance levels where the hybrid followed the same patterns as the parents. Via gene ontology enrichment and microRNA-target association analyses during seed development, we found copies of reproductive, developmental and meiotic genes with transgressive and paternal dominance patterns. Interestingly, maternal dominance was more prominent in hypermethylated and downregulated features during seed formation, contrasting to the general maternal gamete demethylation reported during gametogenesis in angiosperms. Associations between methylation and gene expression allowed identification of putative epialleles with diverse pivotal biological functions during seed formation. Furthermore, most differentially methylated regions, differentially expressed siRNAs and transposable elements were in regions that flanked genes without differential expression. This suggests that differential expression and methylation of epigenomic features may help maintain expression of pivotal genes in a hybrid context. Differential expression and methylation patterns during seed formation in an F1 hybrid provide novel insights into genes and mechanisms with potential roles in early heterosis.
© 2023. The Author(s).
Conflict of interest statement
The authors have no relevant financial or non-financial interests to disclose.
Figures




References
-
- Andrews, S. (2010). FastQC: A quality control tool for high throughput sequence data. https://www.bioinformatics.babraham.ac.uk/projects/fastqc. Accessed 2021
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
