

Obesity-induced inflammation exacerbates clonal hematopoiesis
- PMID: 37071471
- PMCID: PMC10231999
- DOI: 10.1172/JCI163968
Obesity-induced inflammation exacerbates clonal hematopoiesis
Abstract
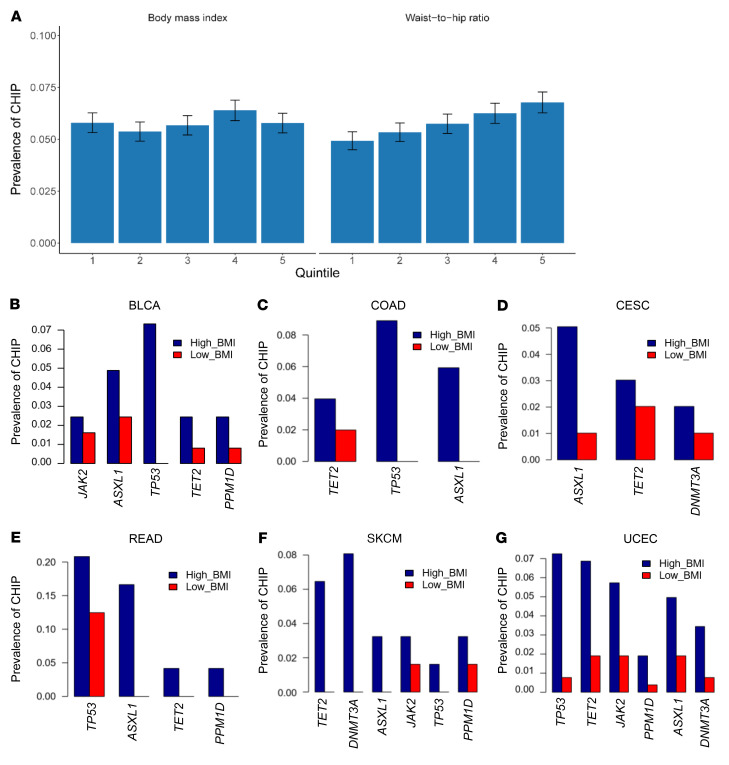
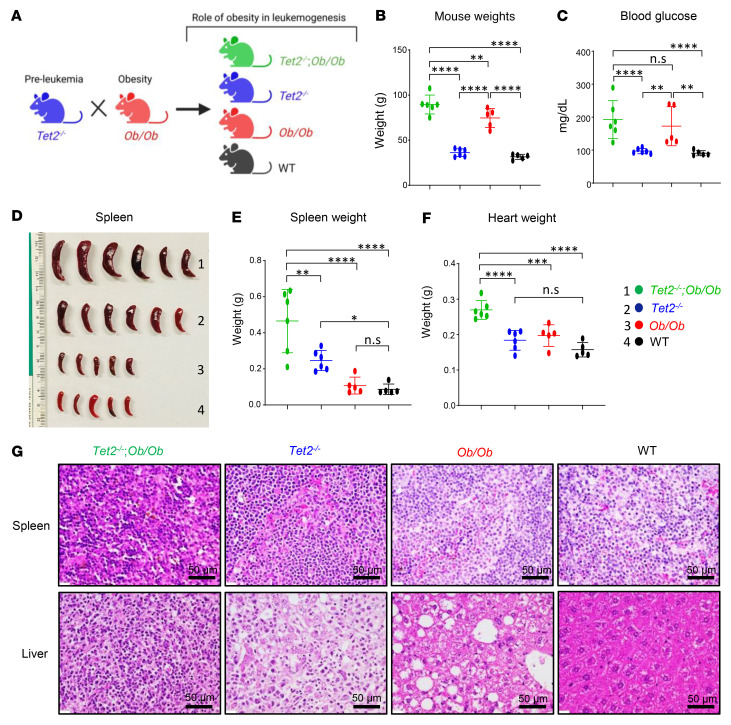
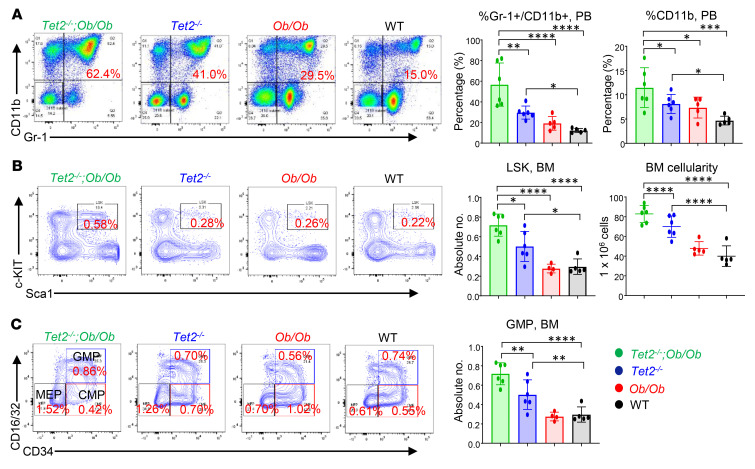
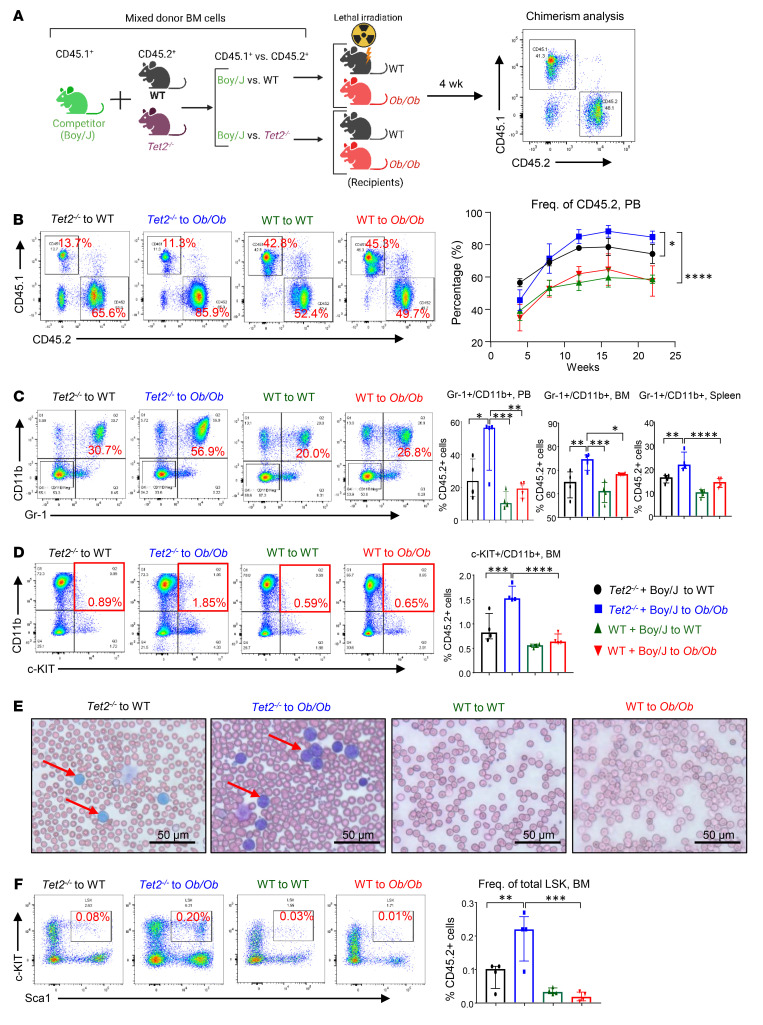
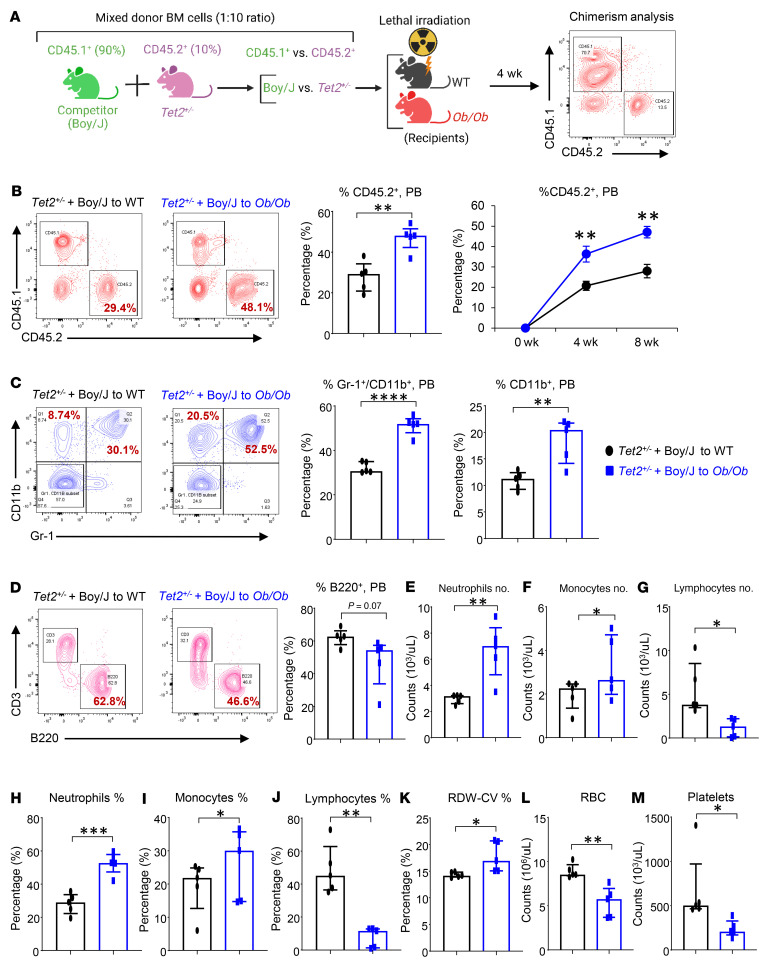
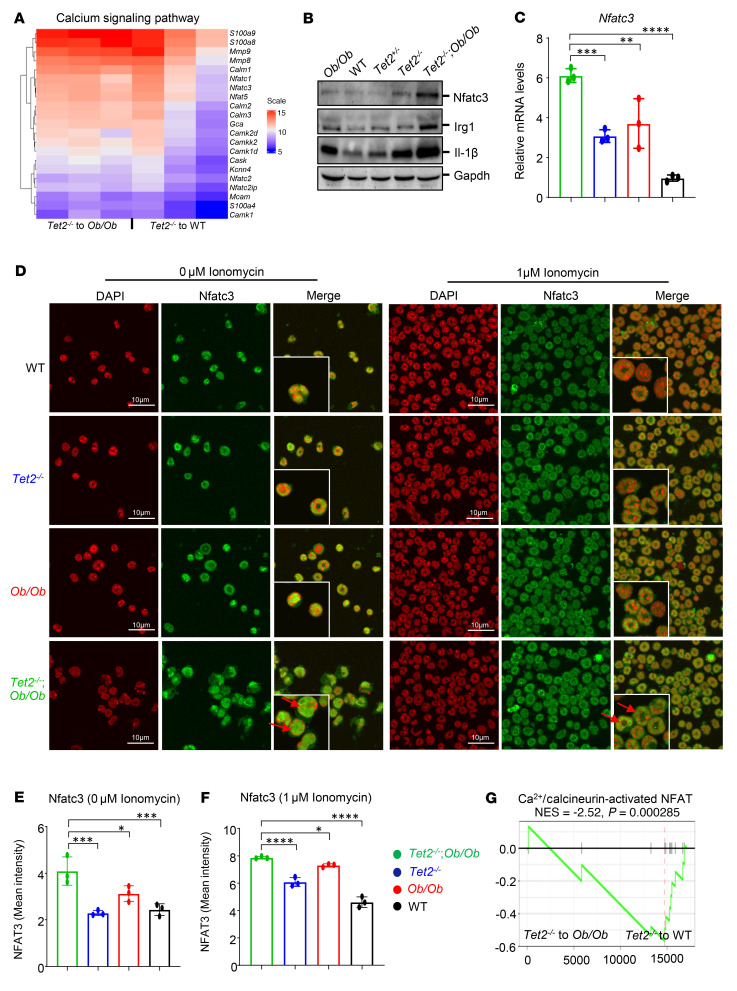
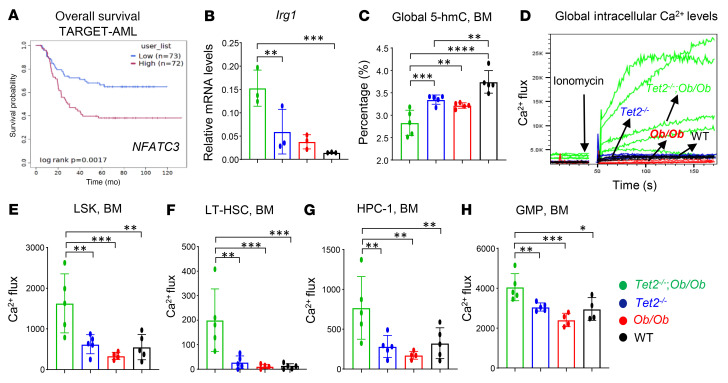
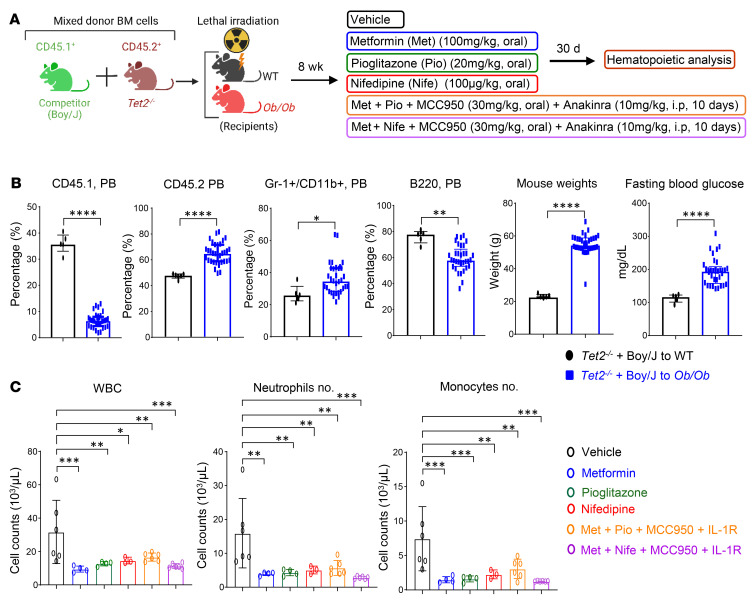
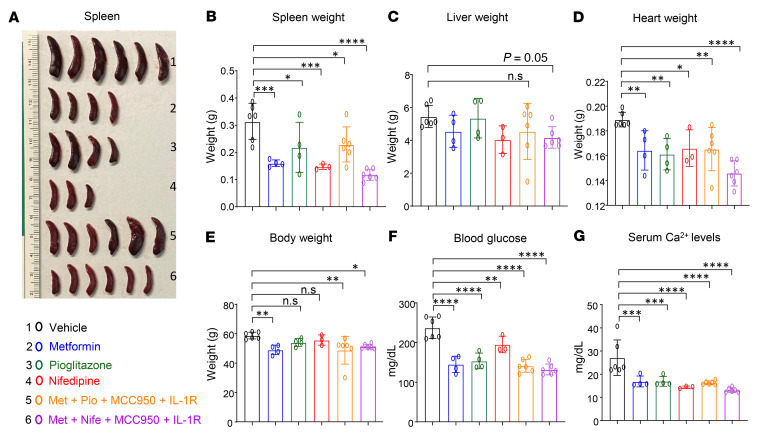
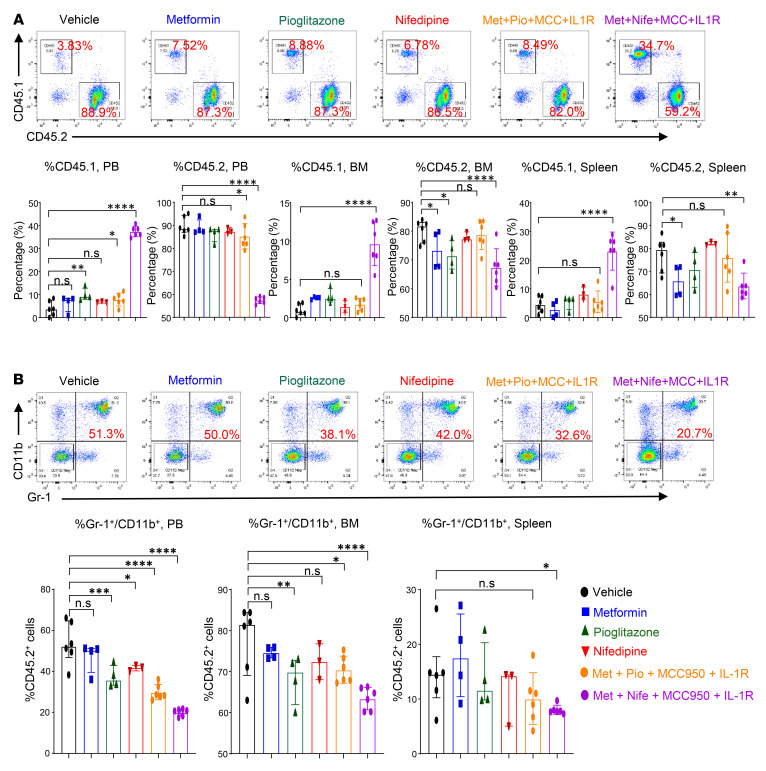
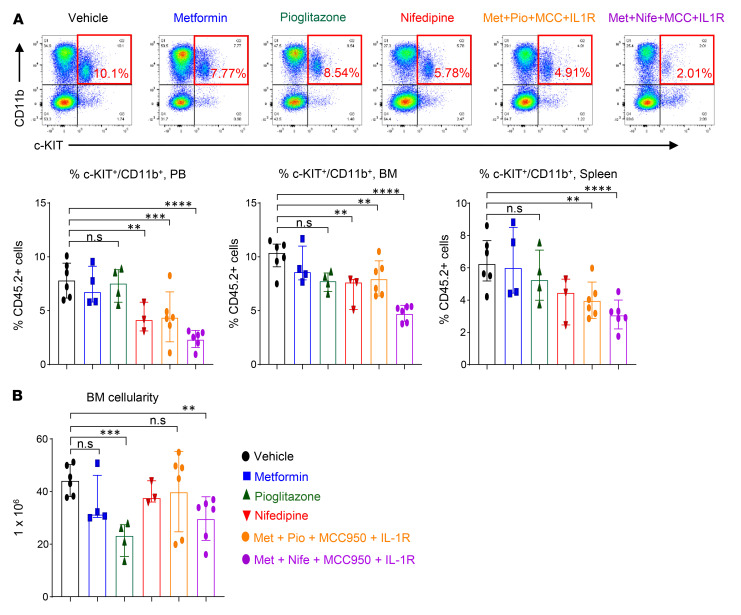
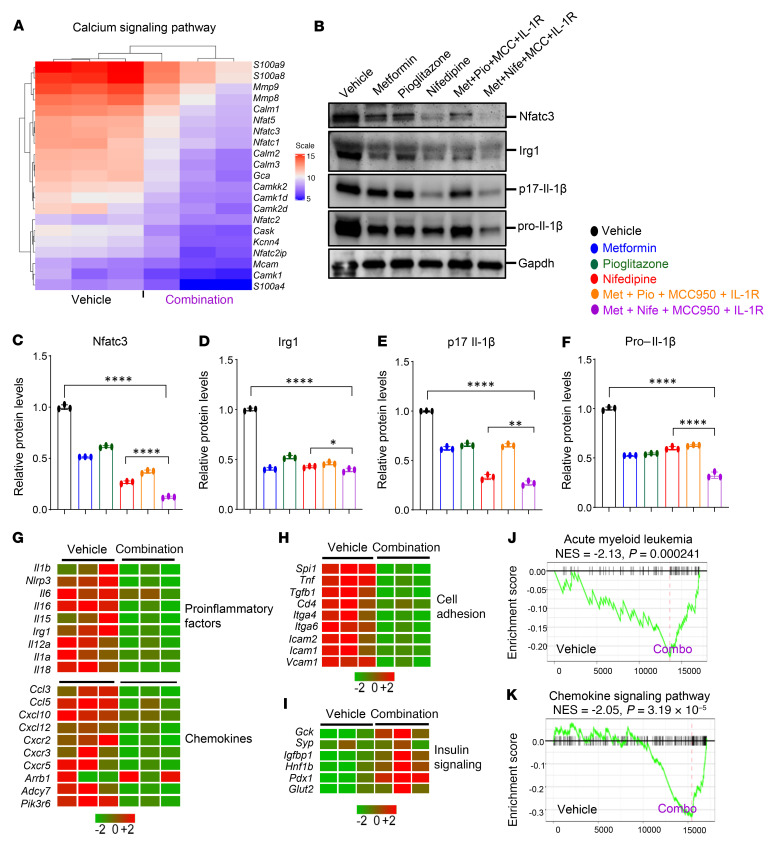
Characterized by the accumulation of somatic mutations in blood cell lineages, clonal hematopoiesis of indeterminate potential (CHIP) is frequent in aging and involves the expansion of mutated hematopoietic stem and progenitor cells (HSC/Ps) that leads to an increased risk of hematologic malignancy. However, the risk factors that contribute to CHIP-associated clonal hematopoiesis (CH) are poorly understood. Obesity induces a proinflammatory state and fatty bone marrow (FBM), which may influence CHIP-associated pathologies. We analyzed exome sequencing and clinical data for 47,466 individuals with validated CHIP in the UK Biobank. CHIP was present in 5.8% of the study population and was associated with a significant increase in the waist-to-hip ratio (WHR). Mouse models of obesity and CHIP driven by heterozygosity of Tet2, Dnmt3a, Asxl1, and Jak2 resulted in exacerbated expansion of mutant HSC/Ps due in part to excessive inflammation. Our results show that obesity is highly associated with CHIP and that a proinflammatory state could potentiate the progression of CHIP to more significant hematologic neoplasia. The calcium channel blockers nifedipine and SKF-96365, either alone or in combination with metformin, MCC950, or anakinra (IL-1 receptor antagonist), suppressed the growth of mutant CHIP cells and partially restored normal hematopoiesis. Targeting CHIP-mutant cells with these drugs could be a potential therapeutic approach to treat CH and its associated abnormalities in individuals with obesity.
Keywords: Cancer; Hematology; Hematopoietic stem cells; Inflammation.
Conflict of interest statement
Figures
References
-
- Köhnke T, Majeti R. Clonal hematopoiesis: from mechanisms to clinical intervention. Cancer Discov. 2021;11(12):2987–2997. doi: 10.1158/2159-8290.CD-21-0901. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
