

SRSF1 haploinsufficiency is responsible for a syndromic developmental disorder associated with intellectual disability
- PMID: 37071997
- PMCID: PMC10183470
- DOI: 10.1016/j.ajhg.2023.03.016
SRSF1 haploinsufficiency is responsible for a syndromic developmental disorder associated with intellectual disability
Abstract
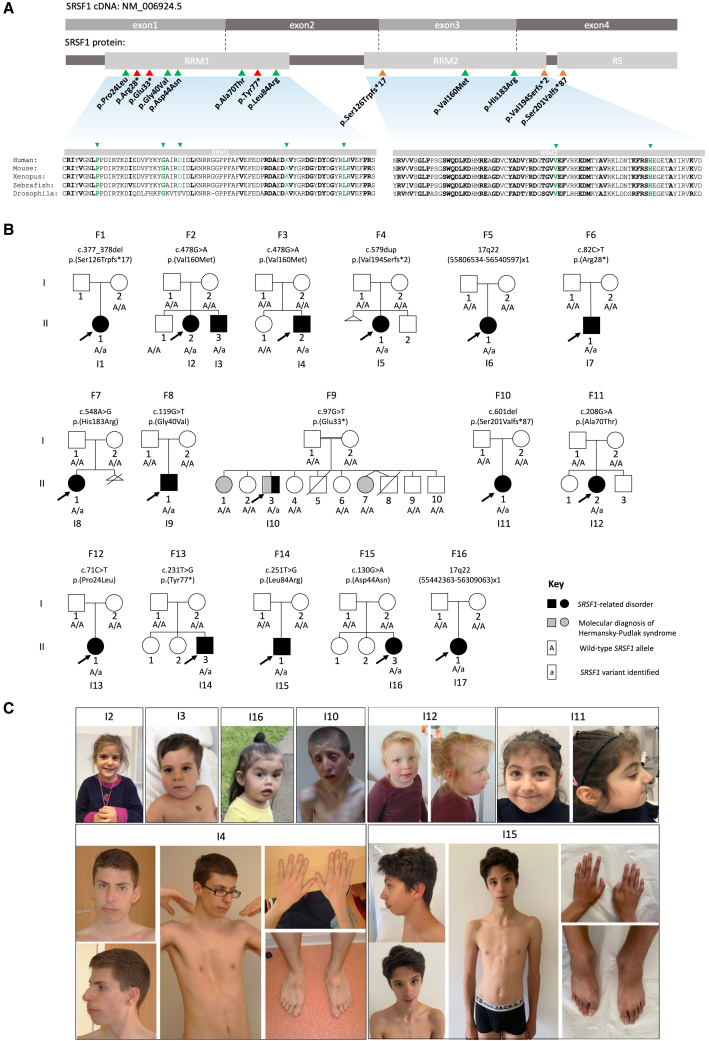
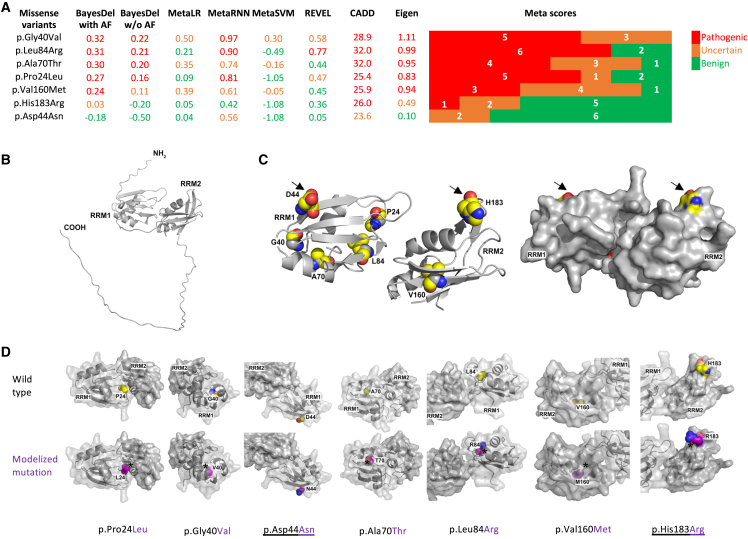
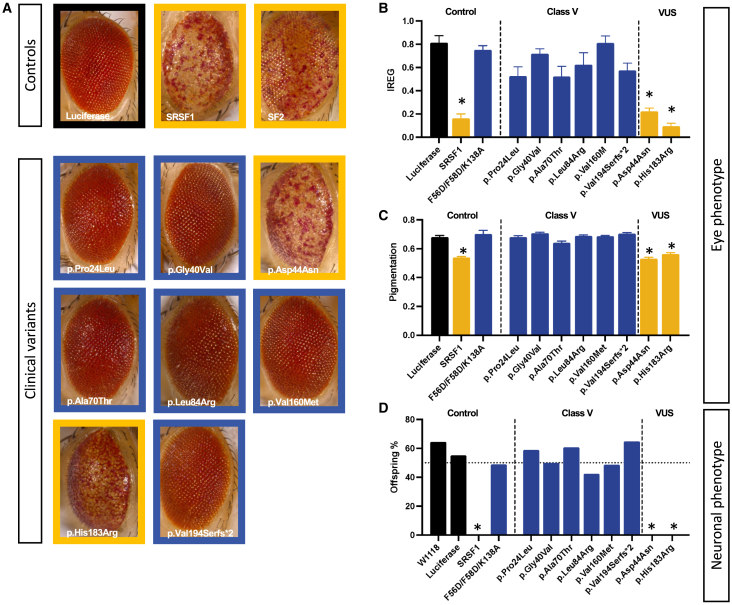
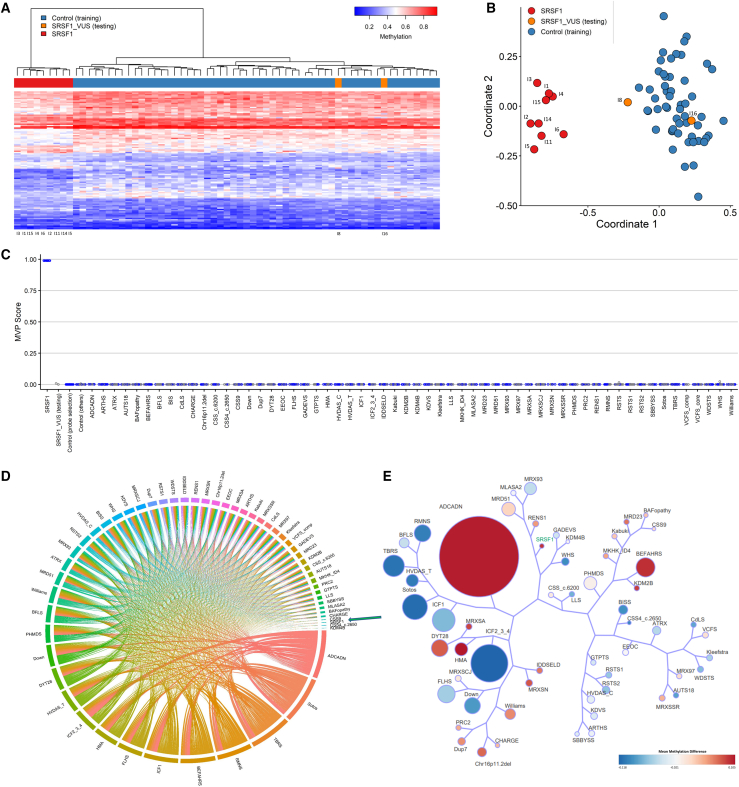
SRSF1 (also known as ASF/SF2) is a non-small nuclear ribonucleoprotein (non-snRNP) that belongs to the arginine/serine (R/S) domain family. It recognizes and binds to mRNA, regulating both constitutive and alternative splicing. The complete loss of this proto-oncogene in mice is embryonically lethal. Through international data sharing, we identified 17 individuals (10 females and 7 males) with a neurodevelopmental disorder (NDD) with heterozygous germline SRSF1 variants, mostly de novo, including three frameshift variants, three nonsense variants, seven missense variants, and two microdeletions within region 17q22 encompassing SRSF1. Only in one family, the de novo origin could not be established. All individuals featured a recurrent phenotype including developmental delay and intellectual disability (DD/ID), hypotonia, neurobehavioral problems, with variable skeletal (66.7%) and cardiac (46%) anomalies. To investigate the functional consequences of SRSF1 variants, we performed in silico structural modeling, developed an in vivo splicing assay in Drosophila, and carried out episignature analysis in blood-derived DNA from affected individuals. We found that all loss-of-function and 5 out of 7 missense variants were pathogenic, leading to a loss of SRSF1 splicing activity in Drosophila, correlating with a detectable and specific DNA methylation episignature. In addition, our orthogonal in silico, in vivo, and epigenetics analyses enabled the separation of clearly pathogenic missense variants from those with uncertain significance. Overall, these results indicated that haploinsufficiency of SRSF1 is responsible for a syndromic NDD with ID due to a partial loss of SRSF1-mediated splicing activity.
Keywords: Drosophila; SRSF1; epigenetic signature; haploinsufficiency; neurodevelopmental disorder; splicing.
Copyright © 2023 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests I.M.W. is an employee of GeneDx, LLC. J.R.L. has stock ownership in 23andMe, is a paid consultant for the Regeneron Genetics Center, and is a co-inventor on multiple United States and European patents related to molecular diagnostics for inherited neuropathies, eye diseases, and bacterial genomic fingerprinting. The Department of Molecular and Human Genetics at Baylor College of Medicine receives revenue from clinical genetic testing conducted at Baylor Genetics (BG) Laboratories. J.R.L. serves on the Scientific Advisory Board of BG.
Figures
References
-
- Wright C.F., McRae J.F., Clayton S., Gallone G., Aitken S., FitzGerald T.W., Jones P., Prigmore E., Rajan D., Lord J., et al. Making new genetic diagnoses with old data: iterative reanalysis and reporting from genome-wide data in 1,133 families with developmental disorders. Genet. Med. 2018;20:1216–1223. doi: 10.1038/gim.2017.246. - DOI - PMC - PubMed
-
- Mitani T., Isikay S., Gezdirici A., Gulec E.Y., Punetha J., Fatih J.M., Herman I., Akay G., Du H., Calame D.G., et al. High prevalence of multilocus pathogenic variation in neurodevelopmental disorders in the Turkish population. Am. J. Hum. Genet. 2021;108:1981–2005. doi: 10.1016/j.ajhg.2021.08.009. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
