

Co-evolution of large inverted repeats and G-quadruplex DNA in fungal mitochondria may facilitate mitogenome stability: the case of Malassezia
- PMID: 37072481
- PMCID: PMC10113387
- DOI: 10.1038/s41598-023-33486-4
Co-evolution of large inverted repeats and G-quadruplex DNA in fungal mitochondria may facilitate mitogenome stability: the case of Malassezia
Abstract
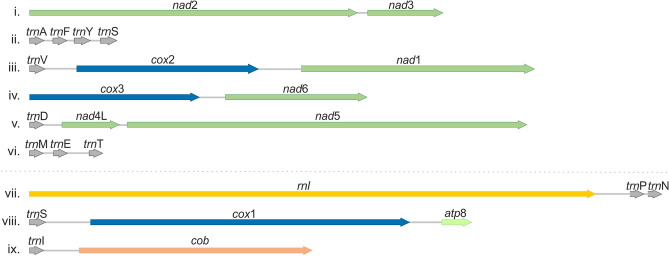
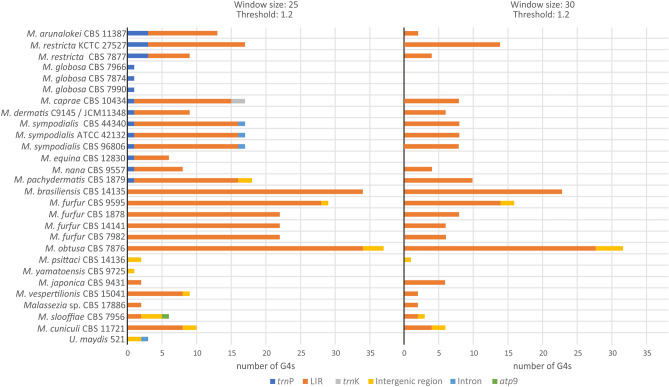
Mitogenomes are essential due to their contribution to cell respiration. Recently they have also been implicated in fungal pathogenicity mechanisms. Members of the basidiomycetous yeast genus Malassezia are an important fungal component of the human skin microbiome, linked to various skin diseases, bloodstream infections, and they are increasingly implicated in gut diseases and certain cancers. In this study, the comparative analysis of Malassezia mitogenomes contributed to phylogenetic tree construction for all species. The mitogenomes presented significant size and gene order diversity which correlates to their phylogeny. Most importantly, they showed the inclusion of large inverted repeats (LIRs) and G-quadruplex (G4) DNA elements, rendering Malassezia mitogenomes a valuable test case for elucidating the evolutionary mechanisms responsible for this genome diversity. Both LIRs and G4s coexist and convergently evolved to provide genome stability through recombination. This mechanism is common in chloroplasts but, hitherto, rarely found in mitogenomes.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures


Similar articles
-
Comparative analysis of Malassezia furfur mitogenomes and the development of a mitochondria-based typing approach.FEMS Yeast Res. 2021 Oct 12;21(7):foab051. doi: 10.1093/femsyr/foab051. FEMS Yeast Res. 2021. PMID: 34562093 Free PMC article.
-
Characterization of G4 DNA formation in mitochondrial DNA and their potential role in mitochondrial genome instability.FEBS J. 2022 Jan;289(1):163-182. doi: 10.1111/febs.16113. Epub 2021 Jul 18. FEBS J. 2022. PMID: 34228888
-
Comparative analysis of the organelle genomes of three Rhodiola species provide insights into their structural dynamics and sequence divergences.BMC Plant Biol. 2023 Mar 22;23(1):156. doi: 10.1186/s12870-023-04159-1. BMC Plant Biol. 2023. PMID: 36944988 Free PMC article.
-
[Phylogenetic relationship of the genus Malassezia based on mitochondrial cytochrome B gene].Nihon Ishinkin Gakkai Zasshi. 2005;46(3):151-6. doi: 10.3314/jjmm.46.151. Nihon Ishinkin Gakkai Zasshi. 2005. PMID: 16094287 Review. Japanese.
-
Malassezia ecology, pathophysiology, and treatment.Med Mycol. 2018 Apr 1;56(suppl_1):S10-S25. doi: 10.1093/mmy/myx134. Med Mycol. 2018. PMID: 29538738 Review.
Cited by
-
Guest edited collection: fungal evolution and diversity.Sci Rep. 2023 Dec 5;13(1):21438. doi: 10.1038/s41598-023-48471-0. Sci Rep. 2023. PMID: 38052958 Free PMC article.
-
Complete Mitochondrial Genomes of Ancyromonads Provide Clues for the Gene Content and Genome Structures of Ancestral Mitochondria.J Eukaryot Microbiol. 2025 May-Jun;72(3):e70012. doi: 10.1111/jeu.70012. J Eukaryot Microbiol. 2025. PMID: 40384044 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
