

Primary cilia as dynamic and diverse signalling hubs in development and disease
- PMID: 37072495
- PMCID: PMC7615029
- DOI: 10.1038/s41576-023-00587-9
Primary cilia as dynamic and diverse signalling hubs in development and disease
Abstract
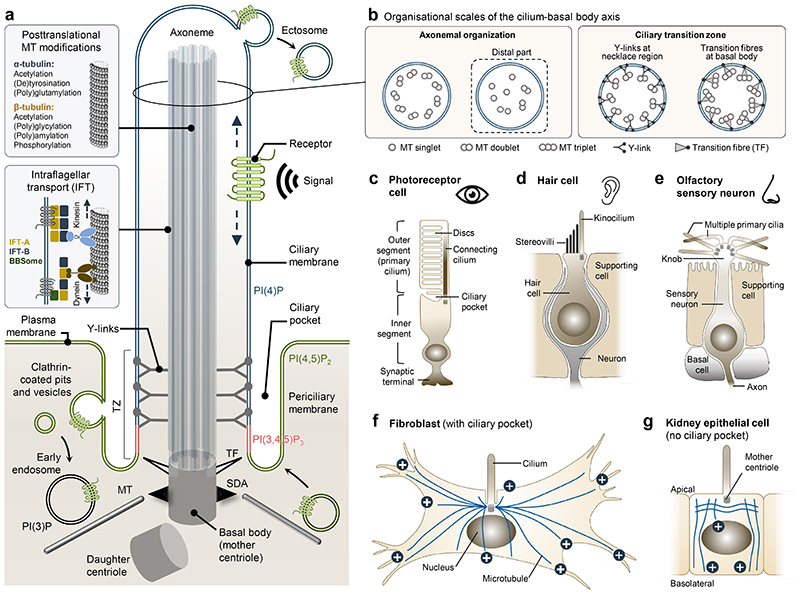
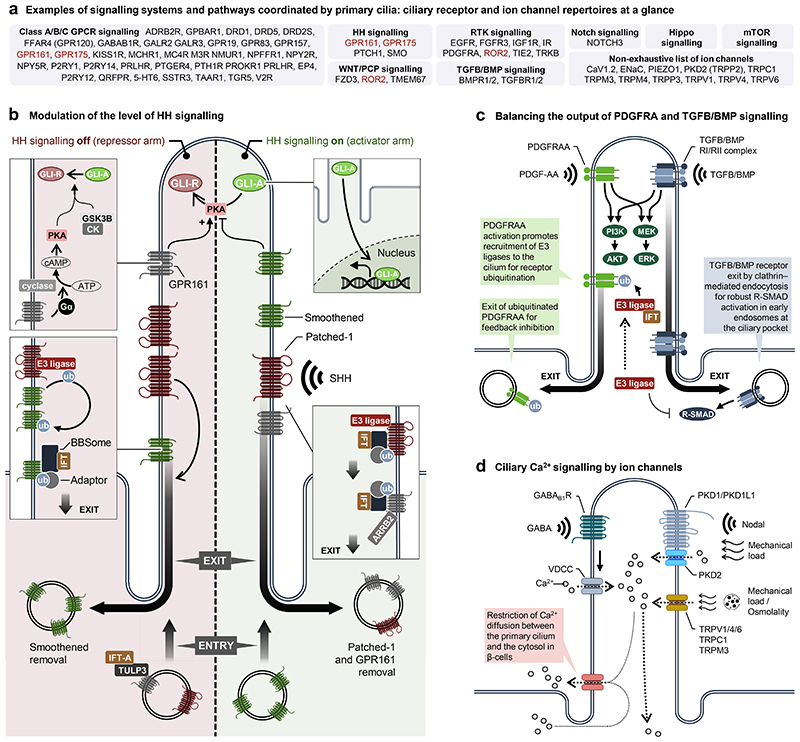
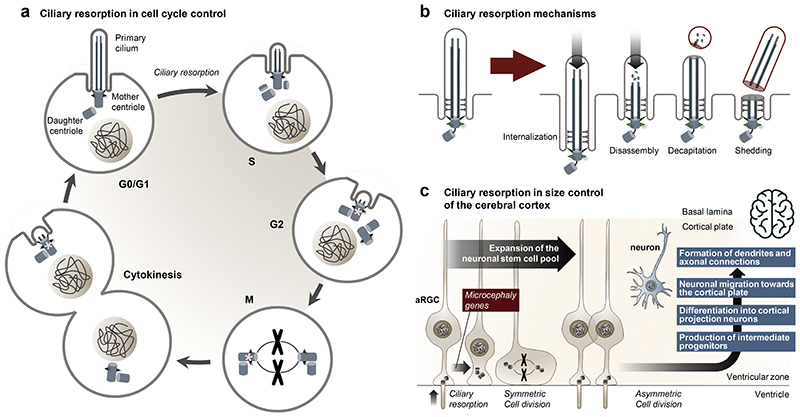
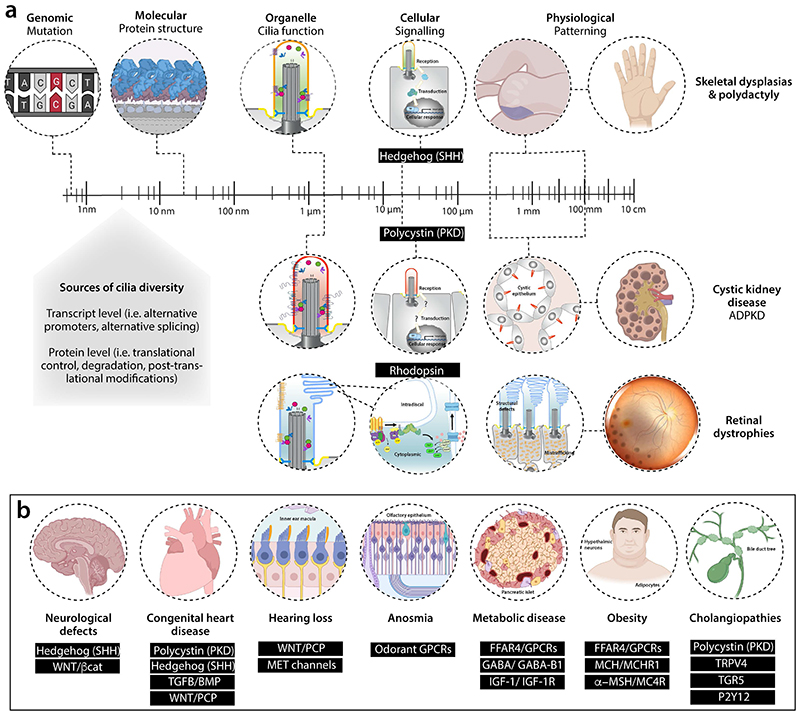
Primary cilia, antenna-like sensory organelles protruding from the surface of most vertebrate cell types, are essential for regulating signalling pathways during development and adult homeostasis. Mutations in genes affecting cilia cause an overlapping spectrum of >30 human diseases and syndromes, the ciliopathies. Given the immense structural and functional diversity of the mammalian cilia repertoire, there is a growing disconnect between patient genotype and associated phenotypes, with variable severity and expressivity characteristic of the ciliopathies as a group. Recent technological developments are rapidly advancing our understanding of the complex mechanisms that control biogenesis and function of primary cilia across a range of cell types and are starting to tackle this diversity. Here, we examine the structural and functional diversity of primary cilia, their dynamic regulation in different cellular and developmental contexts and their disruption in disease.
© 2023. Springer Nature Limited.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
