

POLR1A variants underlie phenotypic heterogeneity in craniofacial, neural, and cardiac anomalies
- PMID: 37075751
- PMCID: PMC10183370
- DOI: 10.1016/j.ajhg.2023.03.014
POLR1A variants underlie phenotypic heterogeneity in craniofacial, neural, and cardiac anomalies
Abstract
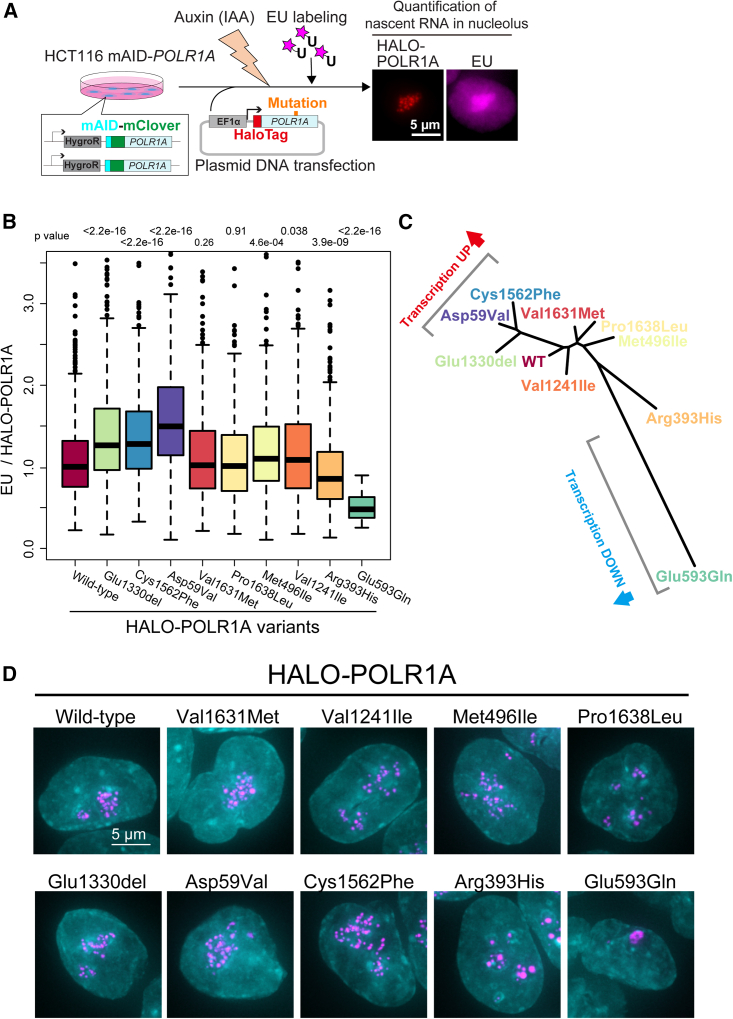
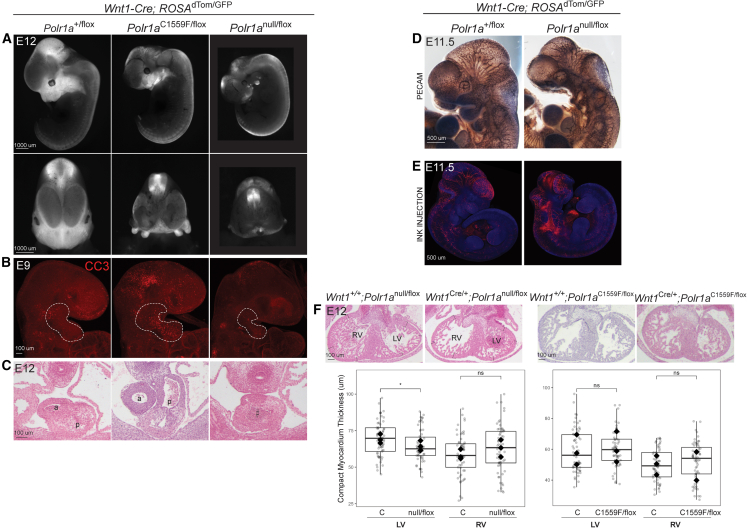
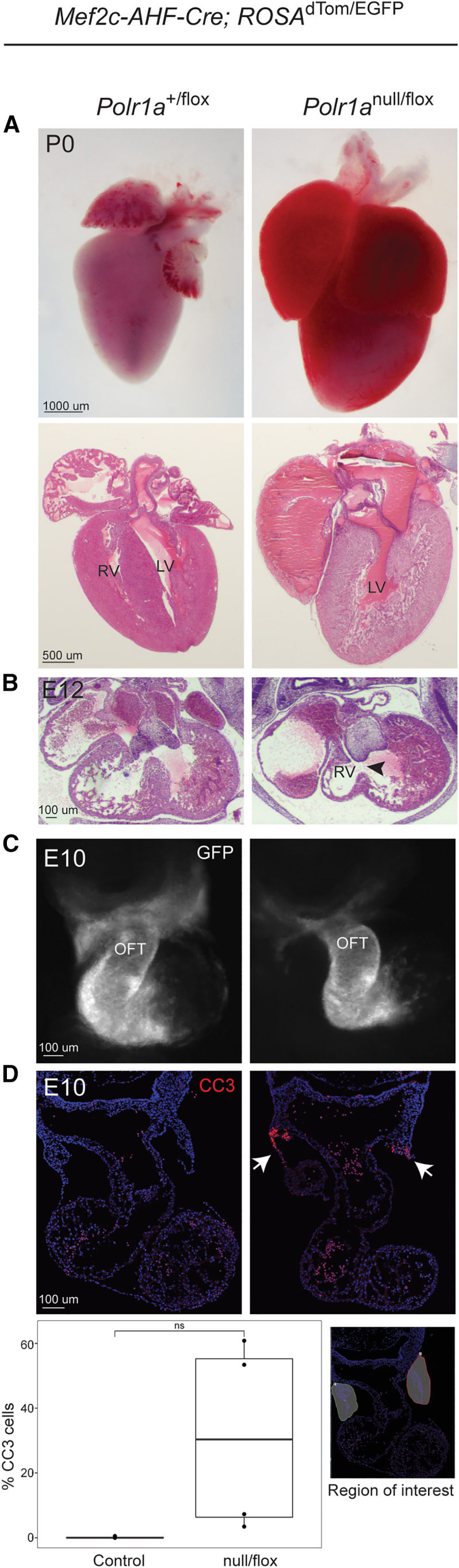
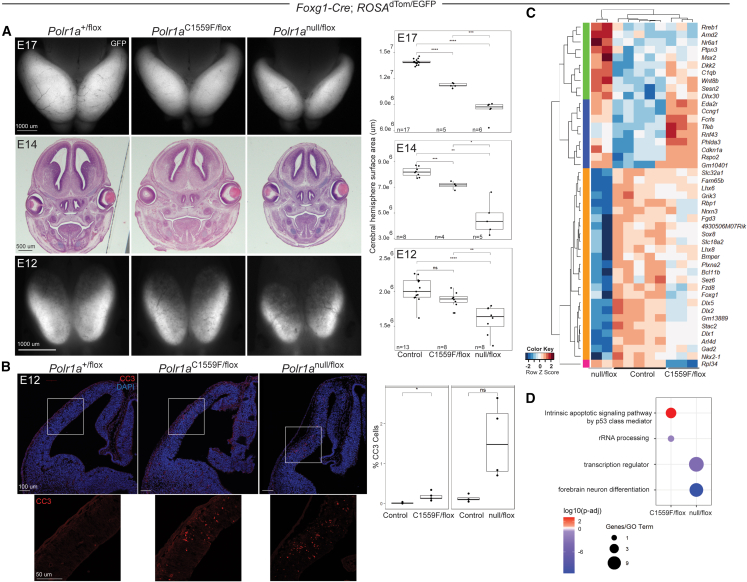
Heterozygous pathogenic variants in POLR1A, which encodes the largest subunit of RNA Polymerase I, were previously identified as the cause of acrofacial dysostosis, Cincinnati-type. The predominant phenotypes observed in the cohort of 3 individuals were craniofacial anomalies reminiscent of Treacher Collins syndrome. We subsequently identified 17 additional individuals with 12 unique heterozygous variants in POLR1A and observed numerous additional phenotypes including neurodevelopmental abnormalities and structural cardiac defects, in combination with highly prevalent craniofacial anomalies and variable limb defects. To understand the pathogenesis of this pleiotropy, we modeled an allelic series of POLR1A variants in vitro and in vivo. In vitro assessments demonstrate variable effects of individual pathogenic variants on ribosomal RNA synthesis and nucleolar morphology, which supports the possibility of variant-specific phenotypic effects in affected individuals. To further explore variant-specific effects in vivo, we used CRISPR-Cas9 gene editing to recapitulate two human variants in mice. Additionally, spatiotemporal requirements for Polr1a in developmental lineages contributing to congenital anomalies in affected individuals were examined via conditional mutagenesis in neural crest cells (face and heart), the second heart field (cardiac outflow tract and right ventricle), and forebrain precursors in mice. Consistent with its ubiquitous role in the essential function of ribosome biogenesis, we observed that loss of Polr1a in any of these lineages causes cell-autonomous apoptosis resulting in embryonic malformations. Altogether, our work greatly expands the phenotype of human POLR1A-related disorders and demonstrates variant-specific effects that provide insights into the underlying pathogenesis of ribosomopathies.
Keywords: RNA Polymerase I; acrofacial dysostosis; congenital heart defect; craniofacial anomalies; developmental delay; epilepsy; limb defects; neural crest cells; ribosomal RNA; ribosomopathies.
Copyright © 2023 American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests A.B., M.J.G.S., K. McWalters, R.P., and R.S. are employees of GeneDx.
Figures

References
-
- Vincent M., Geneviève D., Ostertag A., Marlin S., Lacombe D., Martin-Coignard D., Coubes C., David A., Lyonnet S., Vilain C., et al. Treacher Collins syndrome: a clinical and molecular study based on a large series of patients. Genet. Med. 2016;18:49–56. - PubMed
-
- Dauwerse J.G., Dixon J., Seland S., Ruivenkamp C.A.L., van Haeringen A., Hoefsloot L.H., Peters D.J.M., Boers A.C.d., Daumer-Haas C., Maiwald R., et al. Mutations in genes encoding subunits of RNA polymerases I and III cause Treacher Collins syndrome. Nat. Genet. 2011;43:20–22. - PubMed
-
- Schaefer E., Collet C., Genevieve D., Vincent M., Lohmann D.R., Sanchez E., Bolender C., Eliot M.M., Nürnberg G., Passos-Bueno M.R., et al. Autosomal recessive POLR1D mutation with decrease of TCOF1 mRNA is responsible for Treacher Collins syndrome. Genet. Med. 2014;16:720–724. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
