

Bioactive lipid lysophosphatidic acid species are associated with disease progression in idiopathic pulmonary fibrosis
- PMID: 37075981
- PMCID: PMC10205439
- DOI: 10.1016/j.jlr.2023.100375
Bioactive lipid lysophosphatidic acid species are associated with disease progression in idiopathic pulmonary fibrosis
Abstract
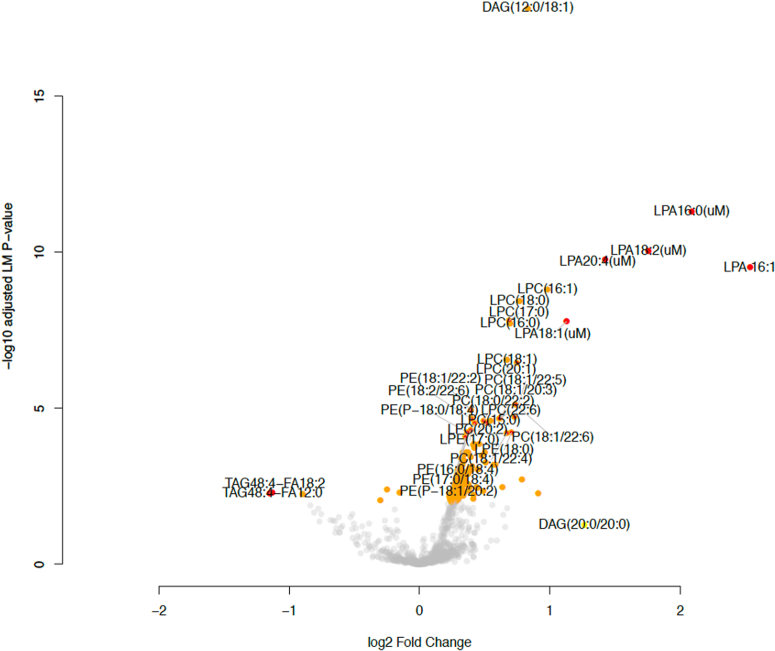
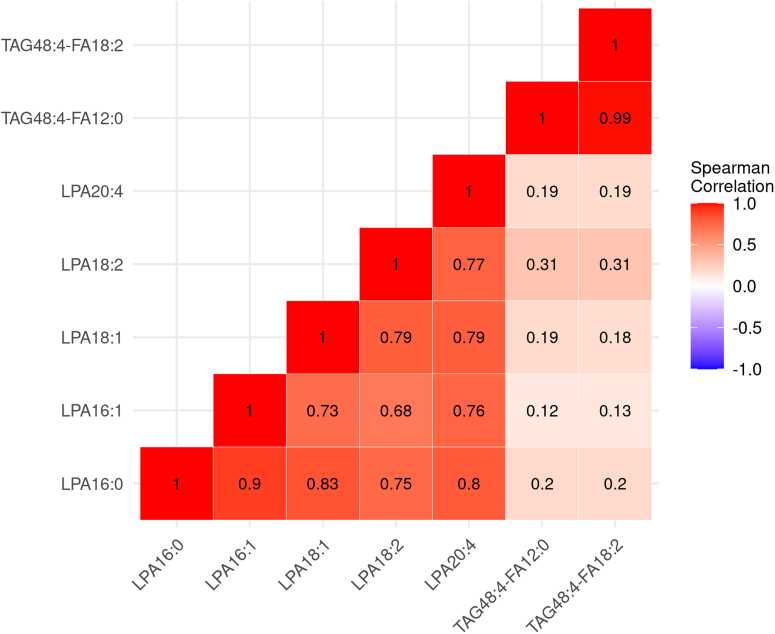
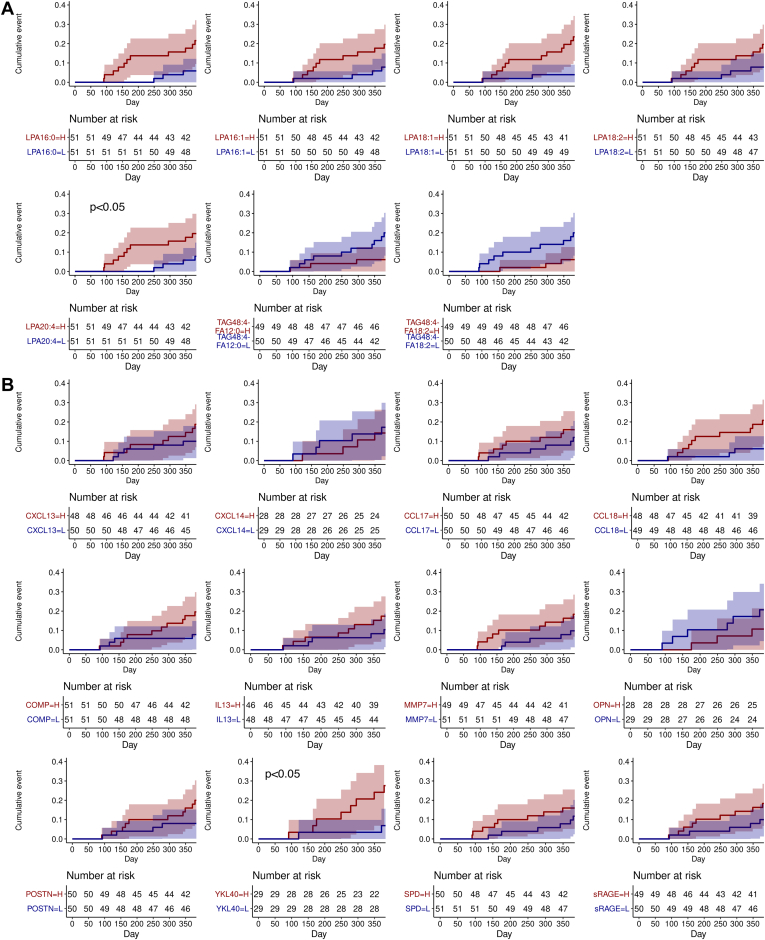
Idiopathic pulmonary fibrosis (IPF) is a progressive disease with significant mortality. Prognostic biomarkers to identify rapid progressors are urgently needed to improve patient management. Since the lysophosphatidic acid (LPA) pathway has been implicated in lung fibrosis in preclinical models and identified as a potential therapeutic target, we aimed to investigate if bioactive lipid LPA species could be prognostic biomarkers that predict IPF disease progression. LPAs and lipidomics were measured in baseline placebo plasma of a randomized IPF-controlled trial. The association of lipids with disease progression indices were assessed using statistical models. Compared to healthy, IPF patients had significantly higher levels of five LPAs (LPA16:0, 16:1, 18:1, 18:2, 20:4) and reduced levels of two triglycerides species (TAG48:4-FA12:0, -FA18:2) (false discovery rate < 0.05, fold change > 2). Patients with higher levels of LPAs had greater declines in diffusion capacity of carbon monoxide over 52 weeks (P < 0.01); additionally, LPA20:4-high (≥median) patients had earlier time to exacerbation compared to LPA20:4-low (<median) patients (hazard ratio (95% CI)): 5.71 (1.17-27.72) (P = 0.031). Higher baseline LPAs were associated with greater increases in fibrosis in lower lungs as quantified by high-resolution computed tomography at week 72 (P < 0.05). Some of these LPAs were positively associated with biomarkers of profibrotic macrophages (CCL17, CCL18, OPN, and YKL40) and lung epithelial damage (SPD and sRAGE) (P < 0.05). In summary, our study established the association of LPAs with IPF disease progression, further supporting the role of the LPA pathway in IPF pathobiology.
Keywords: DLCO; Lysophosphatidic acid; exacerbation; idiopathic pulmonary fibrosis; mortality.
Copyright © 2023 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Conflict of interest The authors declare the following financial interests/personal relationships which may be considered as potential competing interests: M. N., Q. L., W. R. W., G. J., W. S., and G. W. T. are employees of Genentech Inc. S. J. Z and J. L. are employees of Roche.
Figures




References
-
- Adkins J.M., Collard H.R. Idiopathic pulmonary fibrosis. Semin. Respir. Crit. Care Med. 2012;33:433–439. - PubMed
-
- Raghu G., Rochwerg B., Zhang Y., Garcia C.A., Azuma A., Behr J., et al. An official ATS/ERS/JRS/ALAT clinical practice guideline: treatment of idiopathic pulmonary fibrosis. An update of the 2011 clinical practice guideline. Am. J. Respir. Crit. Care Med. 2015;192:e3–e19. - PubMed
-
- Ryerson C.J., Cottin V., Brown K.K., Collard H.R. Acute exacerbation of idiopathic pulmonary fibrosis: shifting the paradigm. Eur. Respir. J. 2015;46:512–520. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
