

Mirusviruses link herpesviruses to giant viruses
- PMID: 37076623
- PMCID: PMC10132985
- DOI: 10.1038/s41586-023-05962-4
Mirusviruses link herpesviruses to giant viruses
Abstract
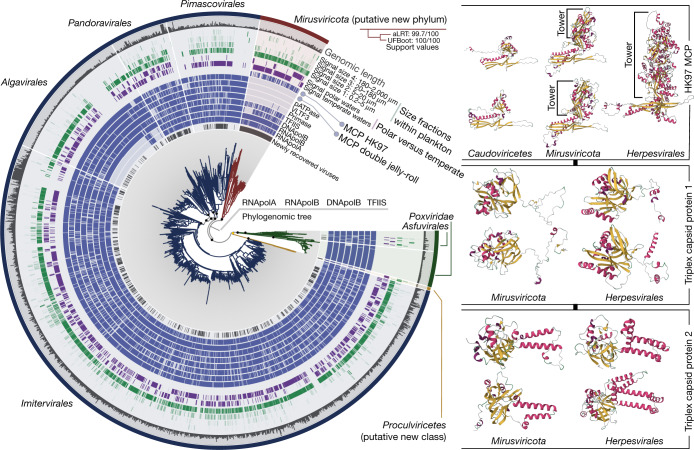
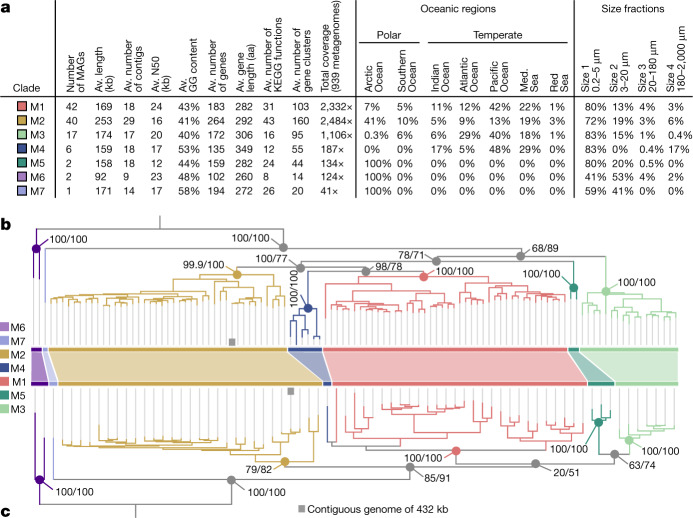
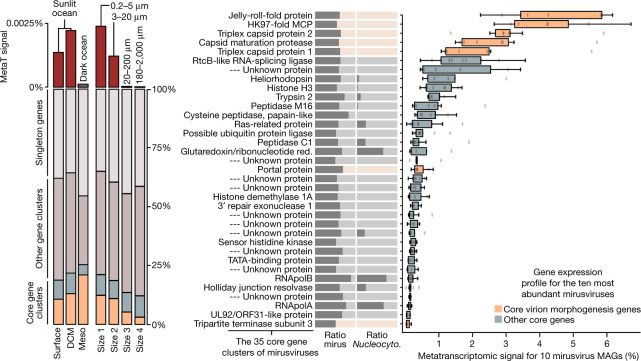
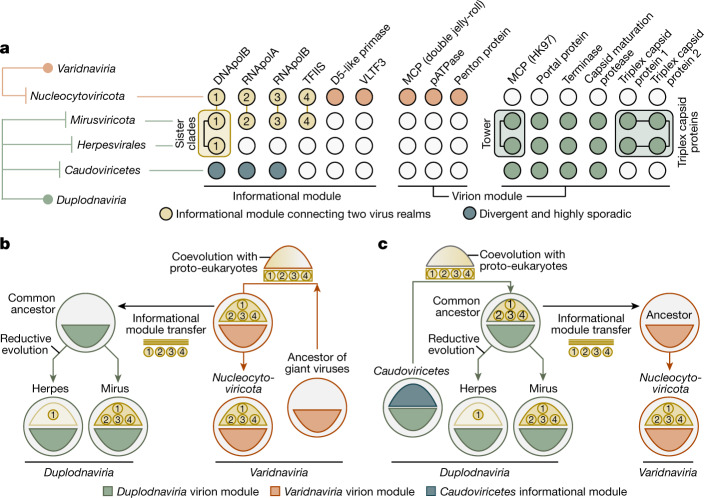
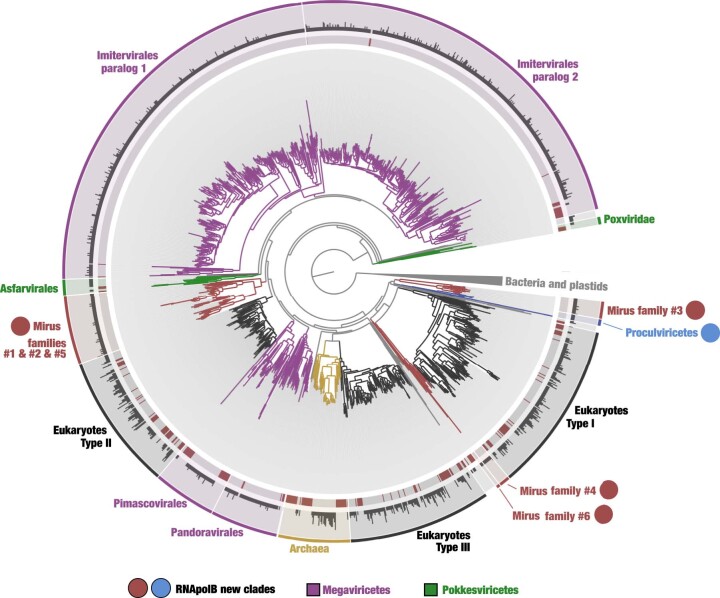
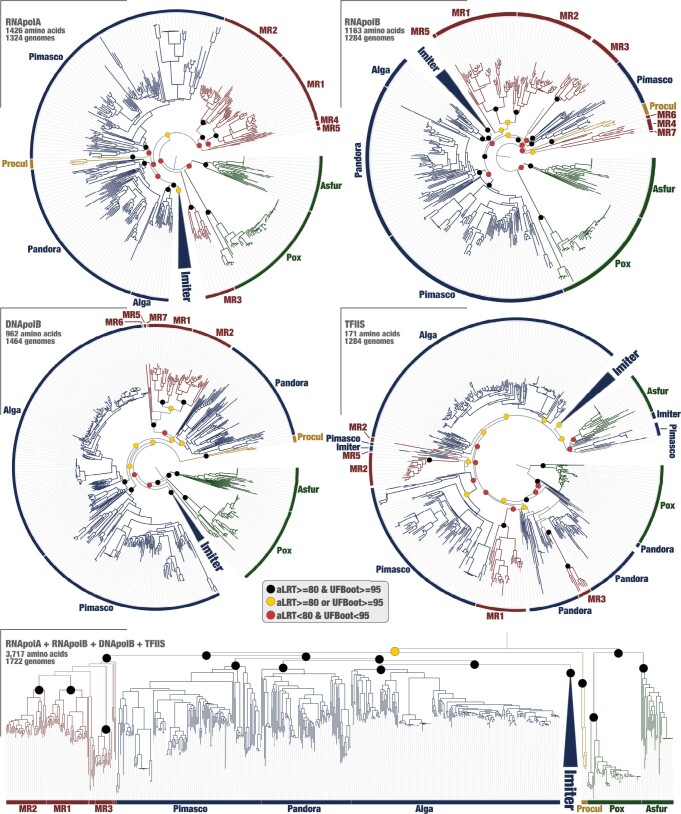
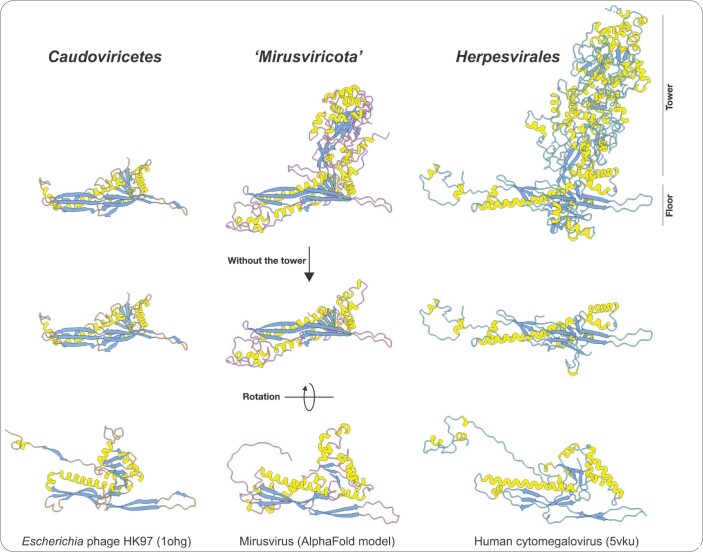
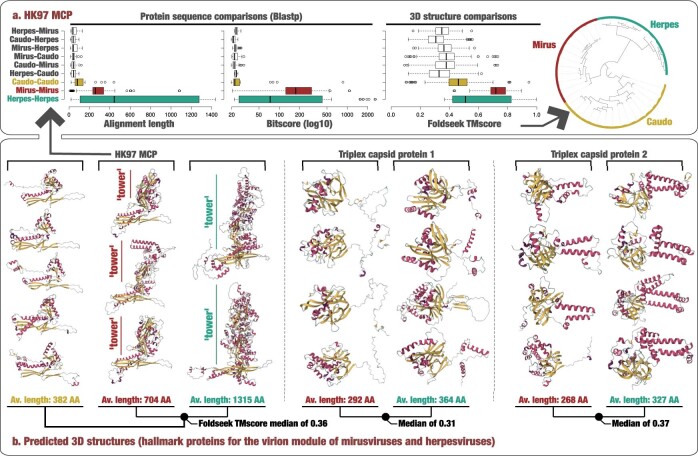
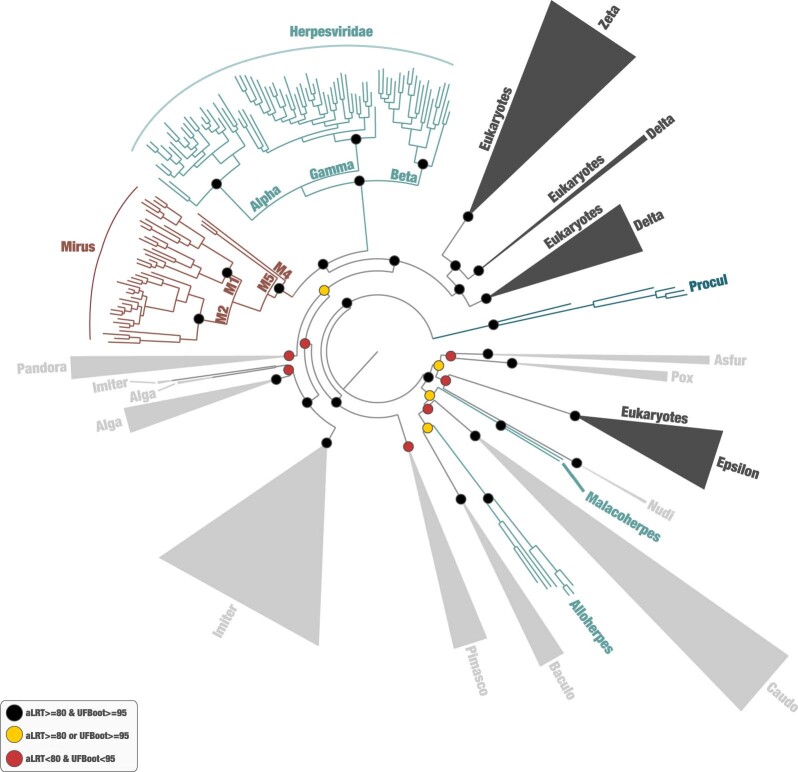
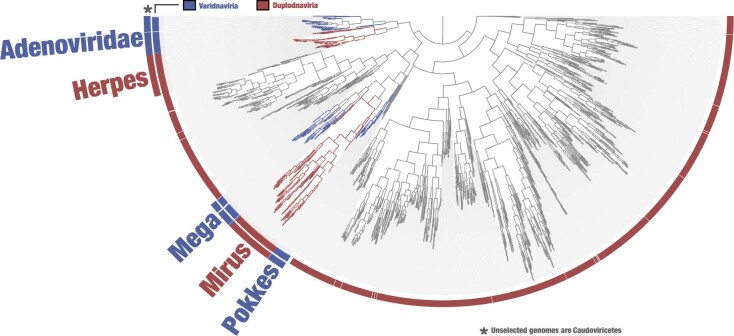
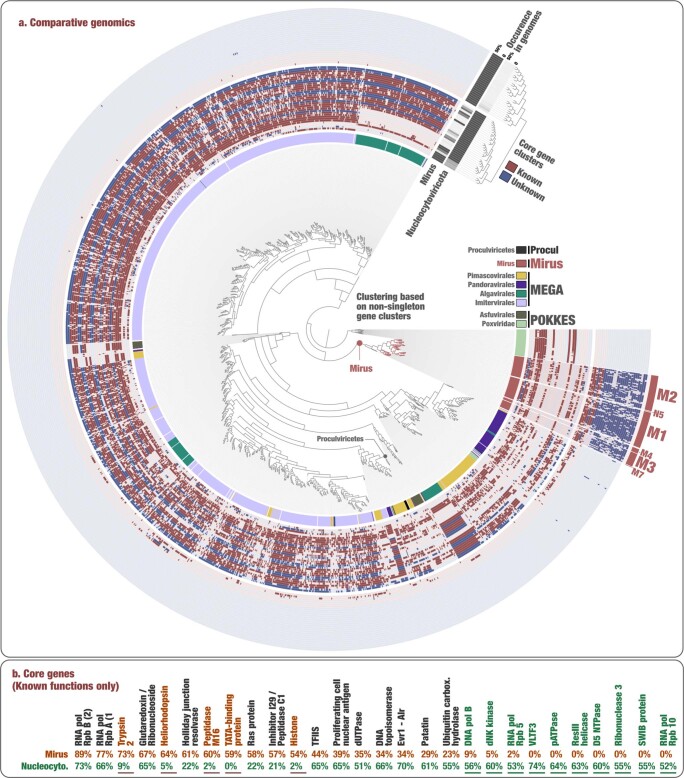
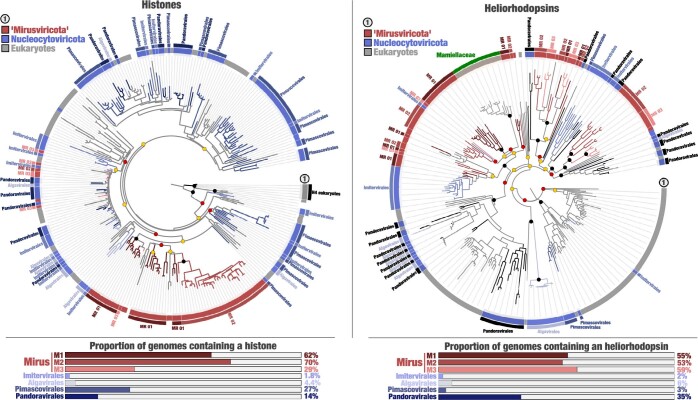
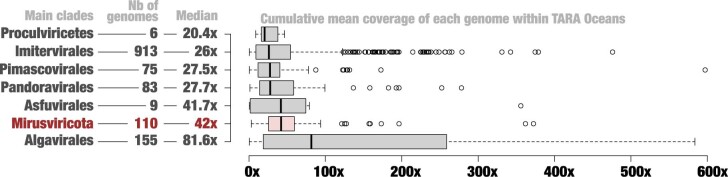
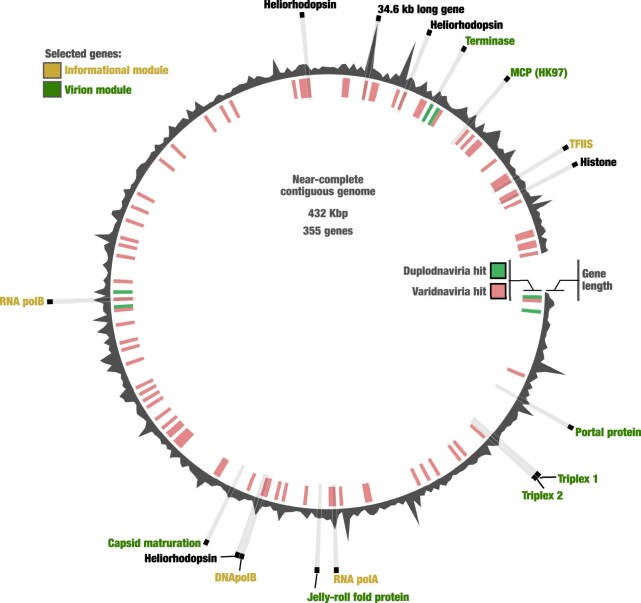
DNA viruses have a major influence on the ecology and evolution of cellular organisms1-4, but their overall diversity and evolutionary trajectories remain elusive5. Here we carried out a phylogeny-guided genome-resolved metagenomic survey of the sunlit oceans and discovered plankton-infecting relatives of herpesviruses that form a putative new phylum dubbed Mirusviricota. The virion morphogenesis module of this large monophyletic clade is typical of viruses from the realm Duplodnaviria6, with multiple components strongly indicating a common ancestry with animal-infecting Herpesvirales. Yet, a substantial fraction of mirusvirus genes, including hallmark transcription machinery genes missing in herpesviruses, are closely related homologues of giant eukaryotic DNA viruses from another viral realm, Varidnaviria. These remarkable chimaeric attributes connecting Mirusviricota to herpesviruses and giant eukaryotic viruses are supported by more than 100 environmental mirusvirus genomes, including a near-complete contiguous genome of 432 kilobases. Moreover, mirusviruses are among the most abundant and active eukaryotic viruses characterized in the sunlit oceans, encoding a diverse array of functions used during the infection of microbial eukaryotes from pole to pole. The prevalence, functional activity, diversification and atypical chimaeric attributes of mirusviruses point to a lasting role of Mirusviricota in the ecology of marine ecosystems and in the evolution of eukaryotic DNA viruses.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Suttle, C. A. Marine viruses — major players in the global ecosystem. Nat. Rev. Microbiol.10.1038/nrmicro1750 (2007). - PubMed
-
- Moniruzzaman, M., Weinheimer, A. R., Martinez-Gutierrez, C. A. & Aylward, F. O. Widespread endogenization of giant viruses shapes genomes of green algae. Nature10.1038/s41586-020-2924-2 (2020). - PubMed
