

Transfer learning in a biomaterial fibrosis model identifies in vivo senescence heterogeneity and contributions to vascularization and matrix production across species and diverse pathologies
- PMID: 37079217
- PMCID: PMC10651581
- DOI: 10.1007/s11357-023-00785-7
Transfer learning in a biomaterial fibrosis model identifies in vivo senescence heterogeneity and contributions to vascularization and matrix production across species and diverse pathologies
Abstract
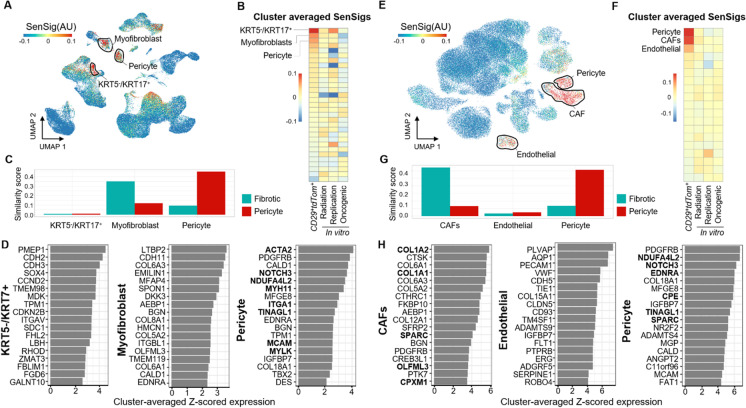
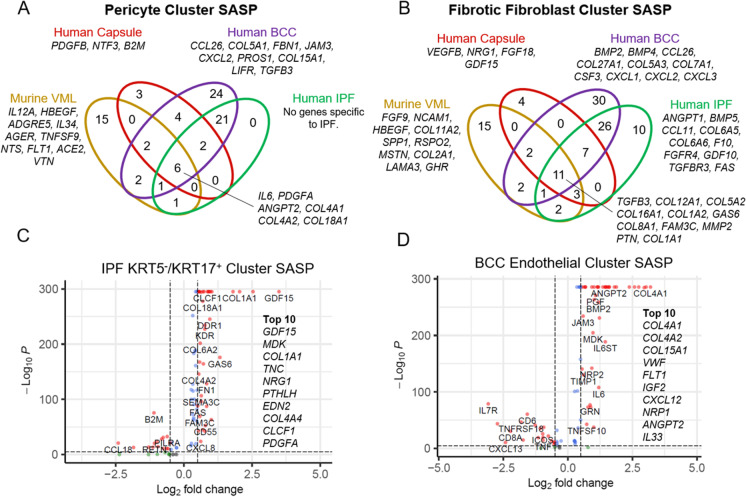
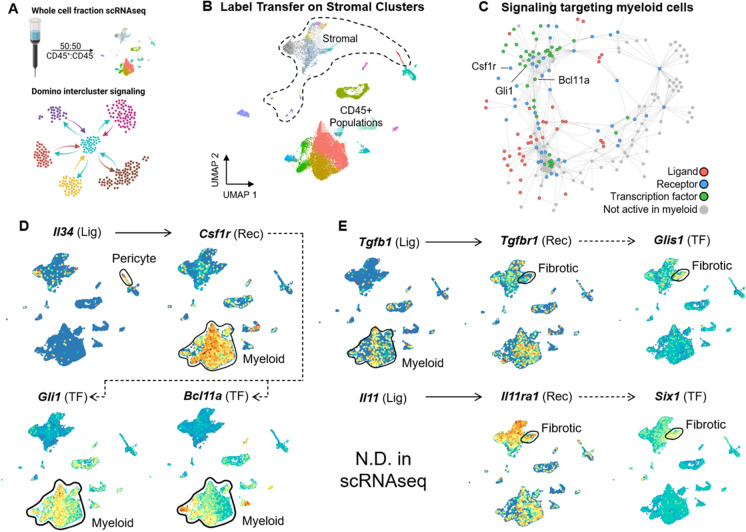
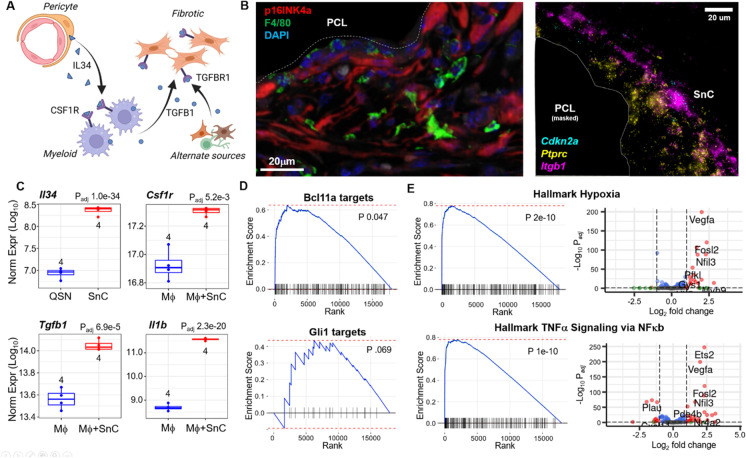
Cellular senescence is a state of permanent growth arrest that plays an important role in wound healing, tissue fibrosis, and tumor suppression. Despite senescent cells' (SnCs) pathological role and therapeutic interest, their phenotype in vivo remains poorly defined. Here, we developed an in vivo-derived senescence signature (SenSig) using a foreign body response-driven fibrosis model in a p16-CreERT2;Ai14 reporter mouse. We identified pericytes and "cartilage-like" fibroblasts as senescent and defined cell type-specific senescence-associated secretory phenotypes (SASPs). Transfer learning and senescence scoring identified these two SnC populations along with endothelial and epithelial SnCs in new and publicly available murine and human data single-cell RNA sequencing (scRNAseq) datasets from diverse pathologies. Signaling analysis uncovered crosstalk between SnCs and myeloid cells via an IL34-CSF1R-TGFβR signaling axis, contributing to tissue balance of vascularization and matrix production. Overall, our study provides a senescence signature and a computational approach that may be broadly applied to identify SnC transcriptional profiles and SASP factors in wound healing, aging, and other pathologies.
Keywords: Fibrosis; RNA sequencing; Senescence.
© 2023. The Author(s), under exclusive licence to American Aging Association.
Conflict of interest statement
J.H.E. holds equity in Unity Biotechnology and Aegeria Soft Tissue and is an advisor for Tessera Therapeutics, HapInScience, and Font Bio. D.M.P. is consultant at Aduro Biotech, Amgen, Astra Zeneca, Bayer, Compugen, DNAtrix, Dynavax Technologies Corporation, Ervaxx, FLX Bio, Immunomic, Janssen, Merck, and Rock Springs Capital. D.M.P. holds equity in Aduro Biotech, DNAtrix, Ervaxx, Five Prime therapeutics, Immunomic, Potenza, and Trieza Therapeutics. D.M.P. is a member of the scientific advisory board for Bristol Myers Squibb, Camden Nexus II, Five Prime Therapeutics, and WindMil. D.M.P. is a member of the board of directors in Dracen Pharmaceuticals. C.C. is the founder and owner of C M Cherry Consulting, LLC. E.J.F. is a member of the scientific advisory board for Resistance Bio and is a consultant for Merck and Mestag Therapeutics. J.M.v.D. is a co-founder of and holds equity in Unity Biotechnology and Cavalry Biosciences. D.J.B. is a shareholder and co-inventor on patent applications licensed to or filed by Unity Biotechnology, a company developing senolytic medicines, including small molecules that selectively eliminate senescent cells. Research in his laboratory has been reviewed by the Mayo Clinic Conflict of Interest Review Board and is being conducted in compliance with Mayo Clinic Conflict of Interest policies.
Figures
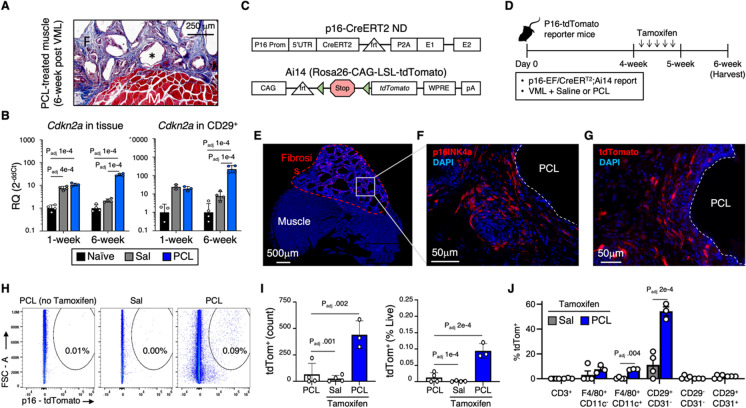
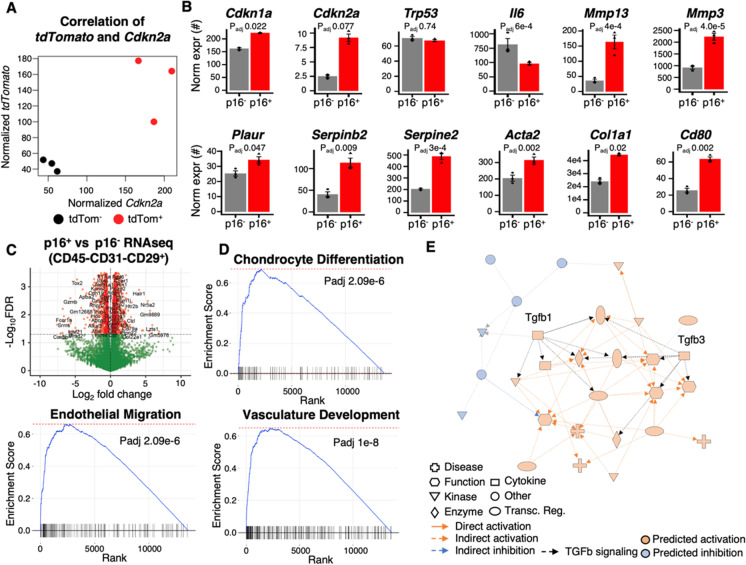
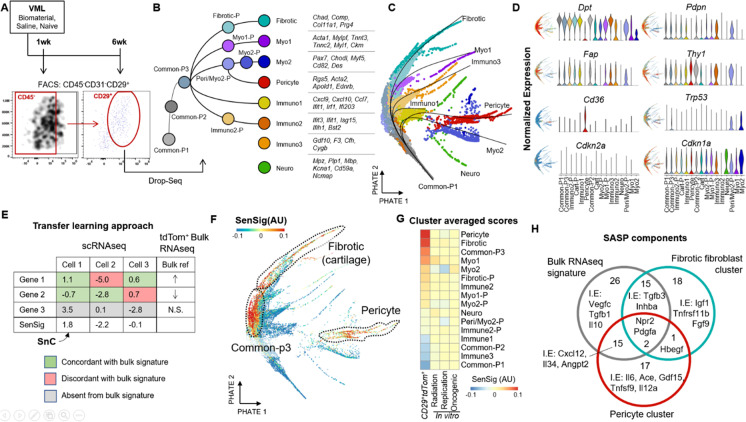
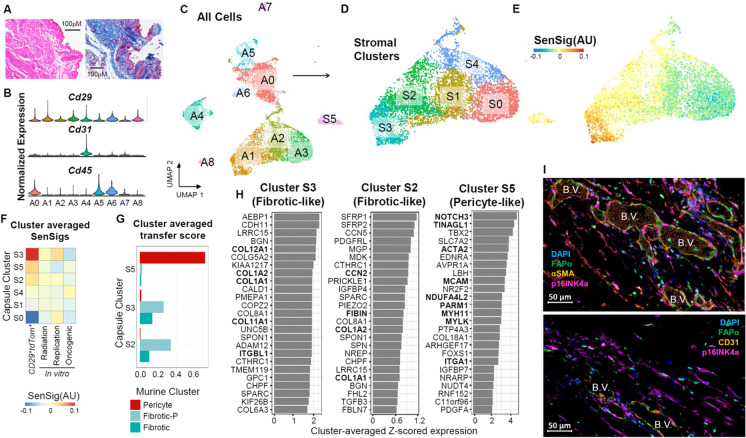
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
